五年前大家做单细胞转录组的时候,三五万左右一个样品的收费让绝大部分课题组都望而却步,一般来说那个时候做少量的几个样品就足够发cns或者子刊级别的文章啦。
但是,很多人珍惜自己的数据,或者说没什么能力把一两个样品的单细胞转录组数据的分析结果编造出来一个生物学故事,卖不出去啊,就只能说是持续贬值了。现在做一个单细胞转录组就五千块钱左右了,时光荏苒岁月如梭啊!现如今做单细胞转录组项目,基本上就跟十年前的普通转录组进化趋势一模一样的,需要有好的实验设计,起码依据自己的生物学背景搞两个分组吧,比如用药前后,转移与否的,生存好坏的不同样品,每个分组起码三五个样品吧。但是这样的话,就有一个很不起眼的风险,就是分组可能会对全局的单细胞转录组表达量没什么特别大的影响,比如取两个分组病人,它们未来是有淋巴结转移这样的结局事件的区别,但是你现在取样他们都是病人的原位肿瘤,这样的话,未来的转移与否并不直接显现在现在的单细胞转录组表达量矩阵的差异上面。
更麻烦的是,有些时候大家有可能自己就把样品分组弄混了,比如2024年1月的文章:《Unveiling inflammatory and prehypertrophic cell populations as key contributors to knee cartilage degeneration in osteoarthritis using multi-omics data integration》,它对应的数据集是:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE255460 有如下所示的19个样品:
GSM8072834 C1_Condition_Control_Rep1
GSM8072835 C2_Condition_Control_Rep2
GSM8072836 C3_Condition_Control_Rep3
GSM8072837 OA1-1_Condition_NWB_Rep1
GSM8072838 OA1-2_Condition_WB_Rep1
GSM8072839 OA2-1_Condition_NWB_Rep2
GSM8072840 OA2-2_Condition_WB_Rep2
GSM8072841 OA3-1_Condition_NWB_Rep3
GSM8072842 OA3-2_Condition_WB_Rep3
GSM8072843 OA4-1_Condition_NWB_Rep4
GSM8072844 OA4-2_Condition_WB_Rep4
GSM8072845 OA5-1_Condition_NWB_Rep5
GSM8072846 OA5-2_Condition_WB_Rep5
GSM8072847 OA6-1_Condition_NWB_Rep6
GSM8072848 OA6-2_Condition_WB_Rep6
GSM8072849 OA7-1_Condition_NWB_Rep7
GSM8072850 OA7-2_Condition_WB_Rep7
GSM8072851 OA8-1_Condition_NWB_Rep8
GSM8072852 OA8-2_Condition_WB_Rep8
分组信息也是很明显:19 cartilage samples from 8 OA donors, and 3 non-OA control donors,而且作者给出来了做出来的降维聚类分群结果文件:
GSE255460_RAW.tar 1.0 Gb (http)(custom) TAR (of MTX, TSV)
GSE255460_metadata.csv.gz 1.9 Mb (ftp)(http) CSV
GSE255460_sc_counts.txt.tar.gz 266.3 Mb (ftp)(http) TAR
我们需要把上面的 GSE255460_RAW.tar 1.0 Gb 文件解压后,整理出下面的结构,然后就可以批量读取到Seurat里面做降维聚类分群,很容易构建成为Seurat对象。仍然是走常规的单细胞转录组降维聚类分群代码,可以看 链接: https://pan.baidu.com/s/1bIBG9RciAzDhkTKKA7hEfQ?pwd=y4eh ,基本上大家只需要读入表达量矩阵文件到r里面就可以使用Seurat包做全部的流程啦:
很容易对比一下自己的降维聚类分群结果,以及文章给出来了的GSE255460_metadata.csv.gz 1.9 Mb里面的降维聚类分群,如下所示的代码:
library(stringr)
phe2=data.table::fread('GSE255460_metadata.csv.gz',
data.table = F)
head(phe2[,c(1,9:12)])
phe2$barcodes = str_split(phe2$V1,'[.]',simplify = T)[,2]
length(unique( phe2$barcodes))
rownames(phe2)=paste(phe2$ID, phe2$barcodes,sep = '_')
table(phe2$ID)
phe=sce.all.int@meta.data
head(phe[,c(1,2)])
phe$orig.ident =gsub('^GSM[0-9]*_','',phe$orig.ident)
table(phe$orig.ident)
phe$barcodes = str_split(rownames(phe),'_',simplify = T)[,3]
length(unique( phe$barcodes))
rownames(phe)=paste(phe$orig.ident, phe$barcodes,sep = '_')
文章给出来了比较详细的单细胞亚群的生物学名字,我不想浪费时间,就不给了:
> head(phe2[,c( 9:12)])
celltype sample trait group
OA1-1_AAACCCACATCAACCA-1 HomC S1 OA NWB
OA1-1_AAACCCACATGTTCAG-1 ProC S1 OA NWB
OA1-1_AAACCCAGTCAGGTGA-1 RegC S1 OA NWB
OA1-1_AAACCCAGTCTGCAAT-1 HomC S1 OA NWB
OA1-1_AAACCCATCAGACTGT-1 ProC S1 OA NWB
OA1-1_AAACCCATCTGCGGAC-1 ProC S1 OA NWB
> head(phe[,c(1,2)])
orig.ident nCount_RNA
C1_AAACCCAGTATGCGTT-1 C1 2716
C1_AAACCCAGTTTCGTGA-1 C1 12216
C1_AAACCCATCTTCTGGC-1 C1 18764
C1_AAACGAAGTCCTCATC-1 C1 27109
C1_AAACGAAGTTTGACAC-1 C1 17225
C1_AAACGAATCTCGTCAC-1 C1 11918
这个时候我们两个人的细胞表型矩阵里面的行名是可以对应的, 因为都是样品名字+细胞的barcode,但是可以看到的是同一个样品在我们两个人的细胞表型矩阵里面的细胞数量居然是差异很大:
> table(phe2$ID)
C1 C2 C3 OA1-1 OA1-2 OA2-1 OA2-2 OA3-1 OA3-2 OA4-1 OA4-2 OA5-1 OA5-2 OA6-1
1345 8183 6979 11731 8994 5263 10457 5004 5245 9807 12006 6458 5063 6003
OA6-2 OA7-1 OA7-2 OA8-1 OA8-2
8519 5935 4690 6577 7637
> table(phe$orig.ident)
C1 C2 C3 OA1-1 OA1-2 OA2-1 OA2-2 OA3-1 OA3-2 OA4-1 OA4-2 OA5-1 OA5-2 OA6-1
1571 10638 7017 13873 11018 5570 10612 6096 8740 7277 8725 5572 5774 10348
OA6-2 OA7-1 OA7-2 OA8-1 OA8-2
12591 6731 5130 6173 5187
这样的问题在于,如果是取交集,就会很可怜:
ids=intersect(rownames(phe),rownames(phe2))
table(phe[ids,'orig.ident'])
g1=phe[ids,]$RNA_snn_res.1
g2=phe2[ids,]$celltype
tmp = table(g1 ,g2 );tmp
gplots::balloonplot(
table(g1 ,g2 )
)
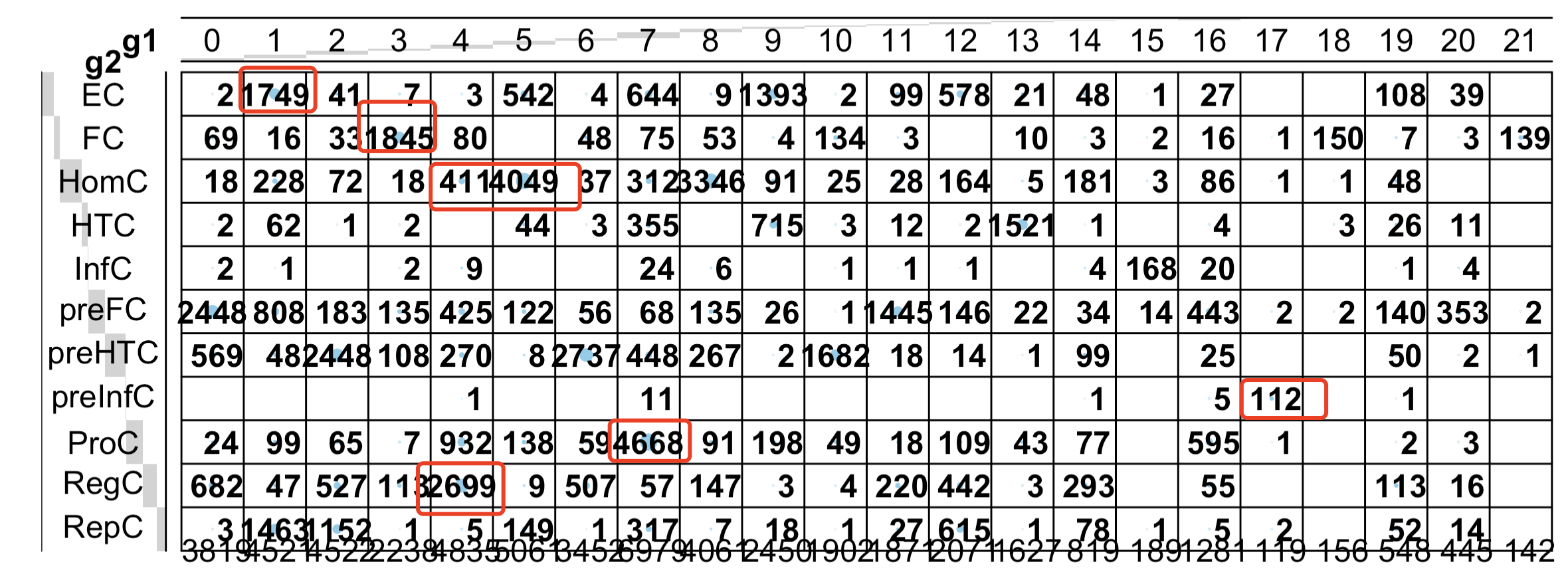
可以看到TMD对应关系并不是很好,这个时候有两个可能性,首先是本来就是单细胞降维聚类分群结果不稳定,亚群直接是没有清晰度界限,分群完全取决于代码运行的参数。另外一个可能性是这交集是错位的。如果是完全错位,理论上它不应该是有一点点比较好的对应关系,既然是有对应成功的,说明是单细胞降维聚类分群结果不稳定。
而且我们两个人的细胞表型矩阵里面的细胞数量都是14万,但是只有5万是可以交集的, 看了看,基本上就前面的7个样品是交集成功了 :
> table(phe[ids,'orig.ident'])
C1 C2 C3 OA1-1 OA1-2 OA2-1 OA2-2 OA3-1 OA3-2 OA4-1 OA4-2 OA5-1 OA5-2 OA6-1
1345 8183 6966 11730 8994 5260 10455 5 14 16 31 11 15 18
OA6-2 OA7-1 OA7-2 OA8-1 OA8-2
25 16 5 8 11
让我想起来了之前遇到了另外一个文章里面的错误,就是不知道10x单细胞转录组样品和fastq文件的对应关系,可以做同样的探索:
list1_barcodes = split(phe$barcodes,phe$orig.ident)
list2_barcodes = split(phe2$barcodes,phe2$ID)
names(list1_barcodes)
tmp = do.call(rbind,
lapply(names(list1_barcodes), function(i){
unlist(lapply(names(list1_barcodes), function(j){
#i=names(list1_barcodes)[1];j=i
sample1_barcodes <- list1_barcodes[[i]]
sample1_length <- as.numeric(length(sample1_barcodes))
sample2_barcodes <- list2_barcodes[[j]]
sample2_length <- as.numeric(length(sample2_barcodes))
both <- intersect(sample1_barcodes,sample2_barcodes)
both_barcodes_length <- as.numeric(length(both))
rate <- as.numeric(2*both_barcodes_length/(sample1_length+sample2_length))
rate
}))
}))
rownames(tmp)=names(list1_barcodes)
colnames(tmp)=names(list1_barcodes)
pheatmap::pheatmap(tmp,
show_rownames = T,show_colnames = T,
cluster_rows = F,cluster_cols = F)
很明显可以看到确实是前面的7个样品是交集成功了:
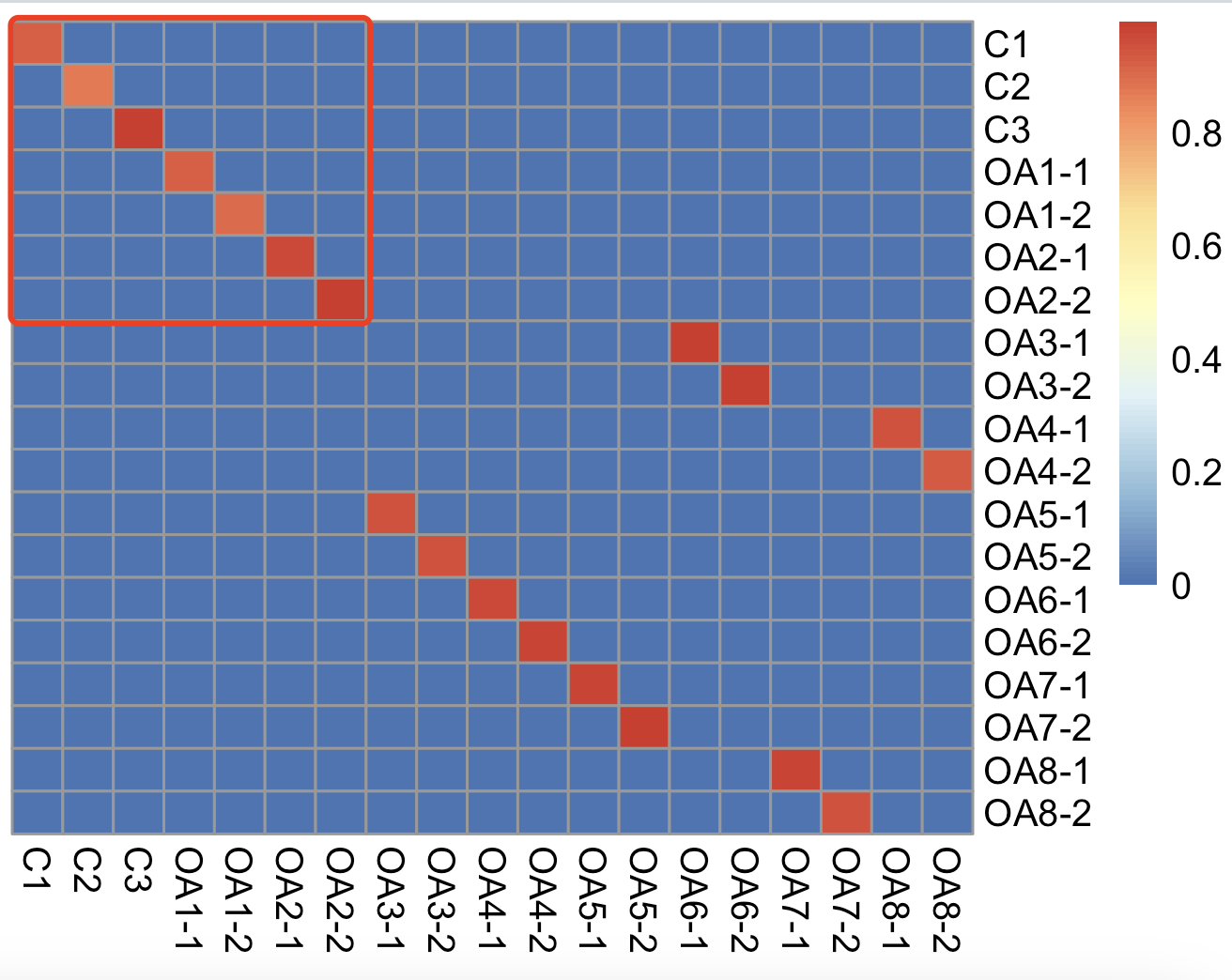
最后的对应关系如下所示:
Var1 Var2 value
C1 C1 0.922496570644719
C2 C2 0.869560597205249
C3 C3 0.995427264932838
OA1-1 OA1-1 0.916263083893142
OA1-2 OA1-2 0.898860683589846
OA2-1 OA2-1 0.971106803286255
OA2-2 OA2-2 0.992453367506763
OA5-1 OA3-1 0.945915279878971
OA5-2 OA3-2 0.951992013794355
OA6-1 OA4-1 0.972860332423716
OA6-2 OA4-2 0.975891368866122
OA7-1 OA5-1 0.976723026764728
OA7-2 OA5-2 0.992249583047189
OA3-1 OA6-1 0.989503264732623
OA3-2 OA6-2 0.986152152500145
OA8-1 OA7-1 0.979517674264949
OA8-2 OA7-2 0.949478586615369
OA4-1 OA8-1 0.949473076367836
OA4-2 OA8-2 0.9333822271116
这样就很有意思了,作者 提供的分组信息也是很明显:19 cartilage samples from 8 OA donors, and 3 non-OA control donors,文章里面的描述如下所示:
- the OA samples were split into weight-bearing (WB) cartilage (n=62 611 chondrocytes) from the medial condyle of the femur
- non-weight-bearing (NWB) cartilage (n=56 778 chondro- cytes) from the intact lateral condyle of the femur to serve as internal controls
文章里面的取样示意图如下所示:
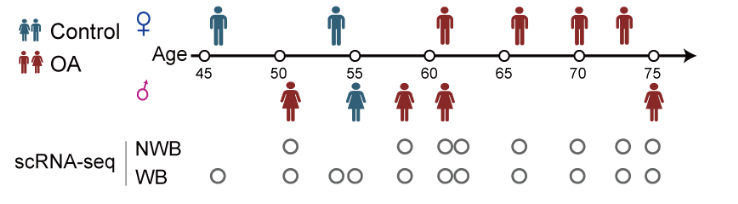
万幸的是虽然说作者把病人的id张冠李戴了,但是它的分组没什么问题,如果它病人id的后缀就是WB和NWB分组。不幸中的万幸?因为文章里面还是有很多关于WB和NWB分组的差异的描述,如果这个弄错了,这个文章就废了。不知道要不要撤稿呢?
不过,这样的小bug其实我并不在意,因为人无完人,数据分析总会有这样的或者那样的bug,我比较在意的生物学bug,比如这样的骨细胞亚群命名:
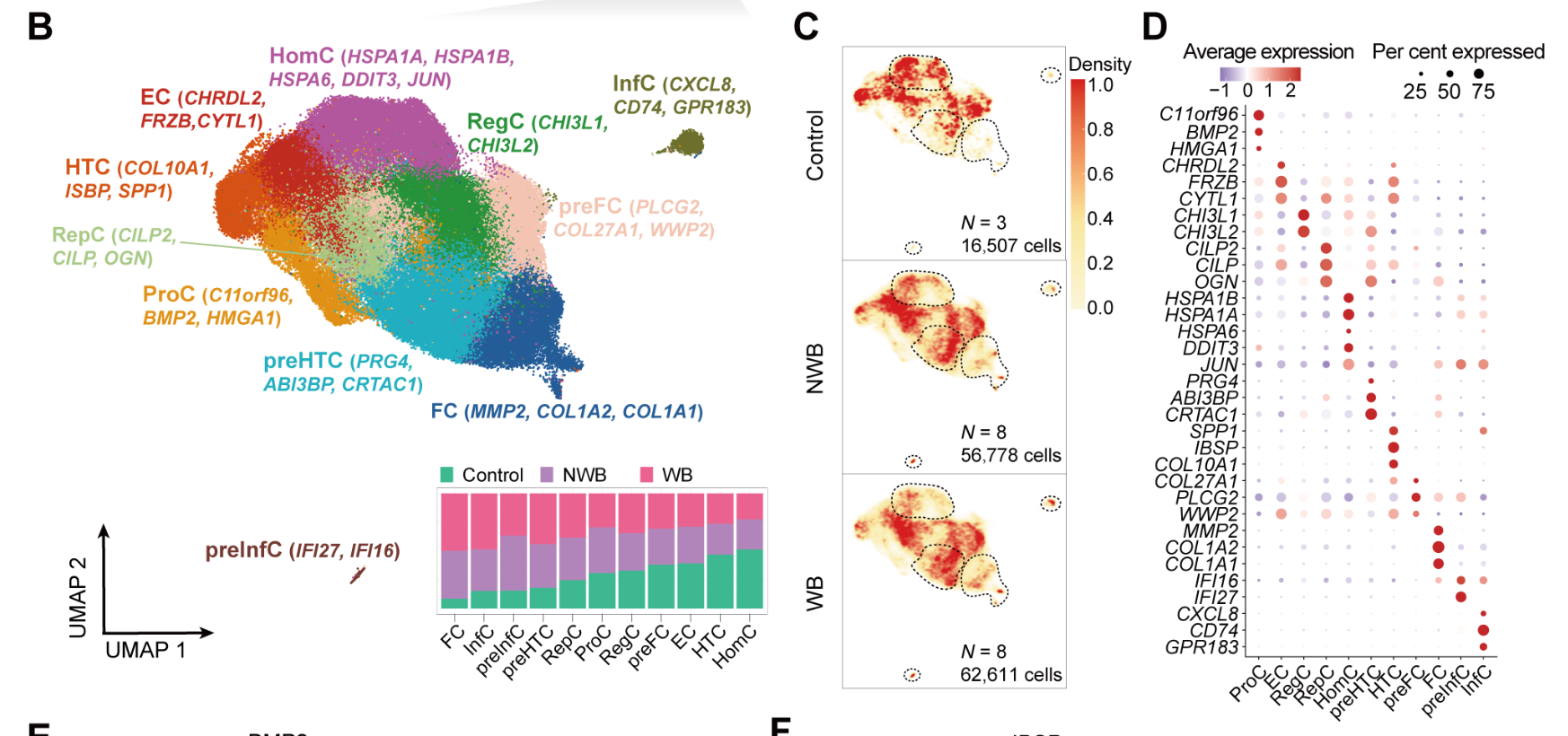
前面我们分享了在单细胞转录组降维聚类分群的第一层次降维聚类分群后的,每个单细胞亚群细分的时候,是有 单细胞亚群的生物学命名的4个规则,如下所示 :
- 第一个规则:已知的生物学亚群
- 第二个规则:顺序编号加上特异性高表达量基因
- 第三个规则:生物学功能注释
- 第四个规则:转录因子等基因集特异性亚群(更多的生物学功能数据库)
一般来说,第一层次降维聚类分群应该是使用第一个规则,但是很明显这个骨的单细胞亚群就很难保证泾渭分明的生物学亚群,仅仅是follow了2017的单细胞文章的命名规则。