在朋友圈刷到了一个(Published: 25 March 2024)的文章:《Assessing GPT-4 for cell type annotation in single-cell RNA-seq analysis》,题目短小精悍,就两个作者,关键是发表在《Nature Methods》杂志,算是生信顶刊了。
GPTCelltype做单细胞亚群注释流程
其实文章所演示的研究者们开发的GPTCelltype做单细胞亚群注释这个过程,我们自己也是在chatGPT界面操作过,就是拿到了各个单细胞亚群的基因后的跟chatGPT普通的对话而已:
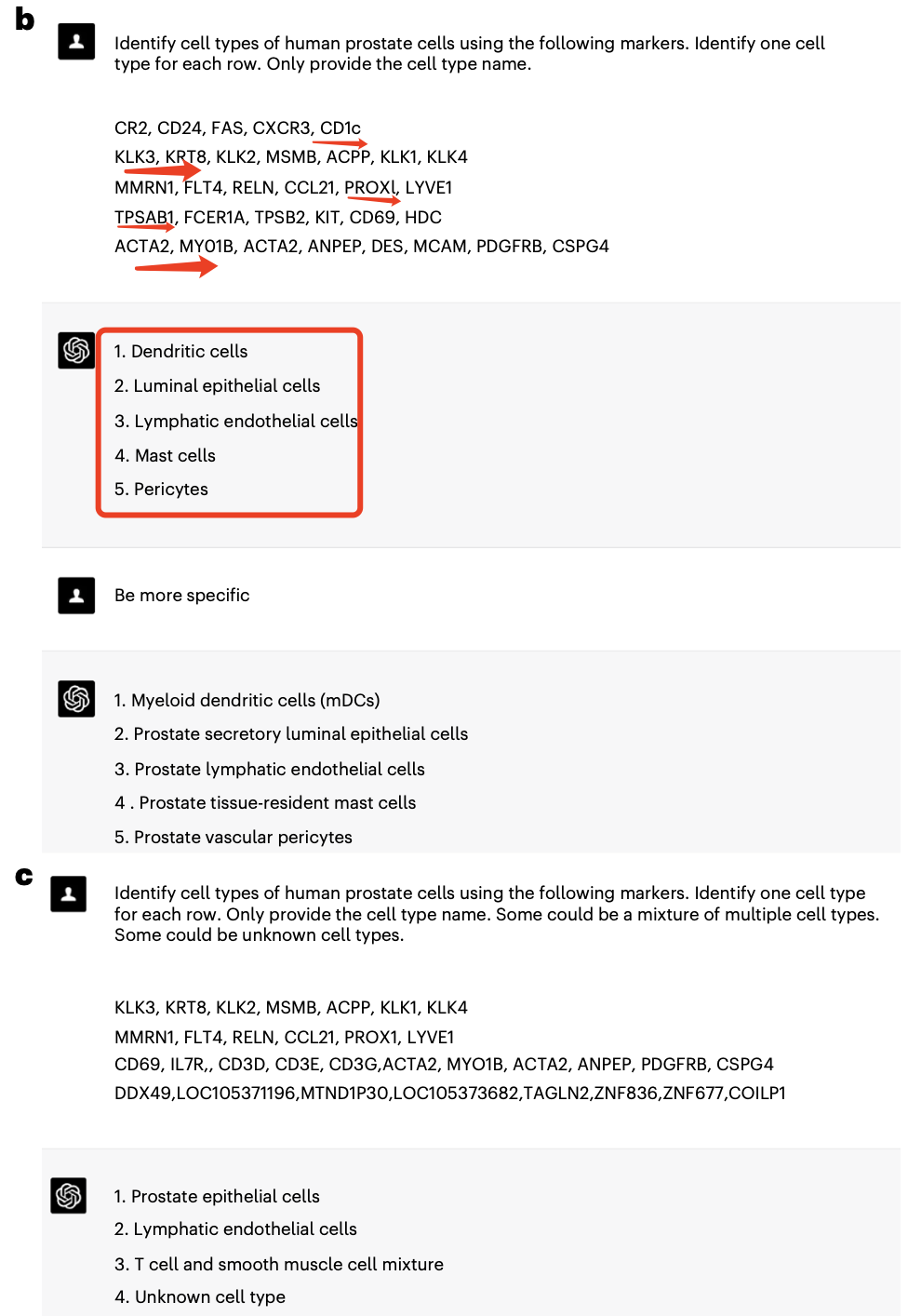
基本上,如果是大家背诵了足够多的基因,是完全没有必要去借助于chatGPT这样的网页工具数据库资源的,人工即可命名。所以文章里面就对比了chatGPT辅助的单细胞亚群注释跟另外的主流的人工注释或者软件(比如 ScType and SingleR)自动化注释的区别:
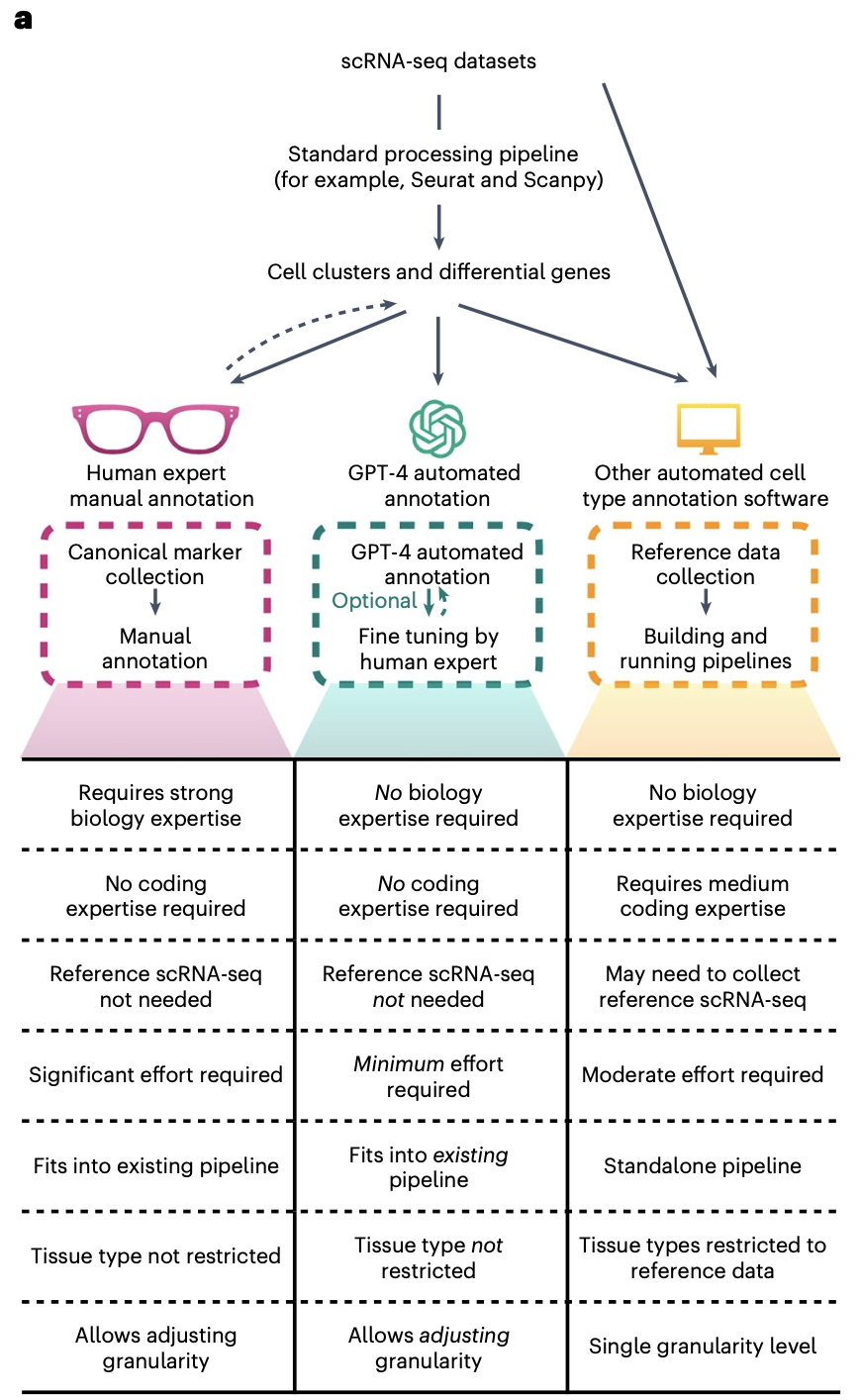
全文就是描述这3种策略的区别,来强调chatGPT辅助的单细胞亚群注释的优缺点而已。如果大家打开文献附带的GPTCelltype包代码地址:
就可以很清晰的看到它的核心就一句话代码而已:res <- gptcelltype(markers, model = 'gpt-4')
要实现这句话,首先是需要安装和加载它的包:
install.packages("openai")
remotes::install_github("Winnie09/GPTCelltype")
# IMPORTANT! Assign your OpenAI API key. See Vignette for details
Sys.setenv(OPENAI_API_KEY = 'your_openai_API_key')
# Load packages
library(GPTCelltype)
library(openai)
麻烦的是这个时候需要打开自己的r运行代码的时候设置一下自己的OPENAI_API_KEY值哦,需要登录chatGPT官网注册账号并且获取 OPENAI_API_KEY 即可。接下来就是自己的单细胞转录组数据的降维聚类分群后的FindAllMarkers拿到每个亚群的基因列表,就可以运行它的核心就一句话代码而已:res <- gptcelltype(markers, model = 'gpt-4'),把命名好的亚群给回去我们的seurat对象即可,如下所示:
# Assume you have already run the Seurat pipeline https://satijalab.org/seurat/
# "obj" is the Seurat object; "markers" is the output from FindAllMarkers(obj)
# Cell type annotation by GPT-4
res <- gptcelltype(markers, model = 'gpt-4')
# Assign cell type annotation back to Seurat object
obj@meta.data$celltype <- as.factor(res[as.character(Idents(obj))])
# Visualize cell type annotation on UMAP
DimPlot(obj,group.by='celltype')
而且如果你打开这个GPTCelltype包源代码,发现其实确实是很简单的,就是把在r里面的各个单细胞亚群的基因组合起来后通过api接口给到chatGPT而已,并不需要你去复制粘贴基因列表去chatGPT官网啦:
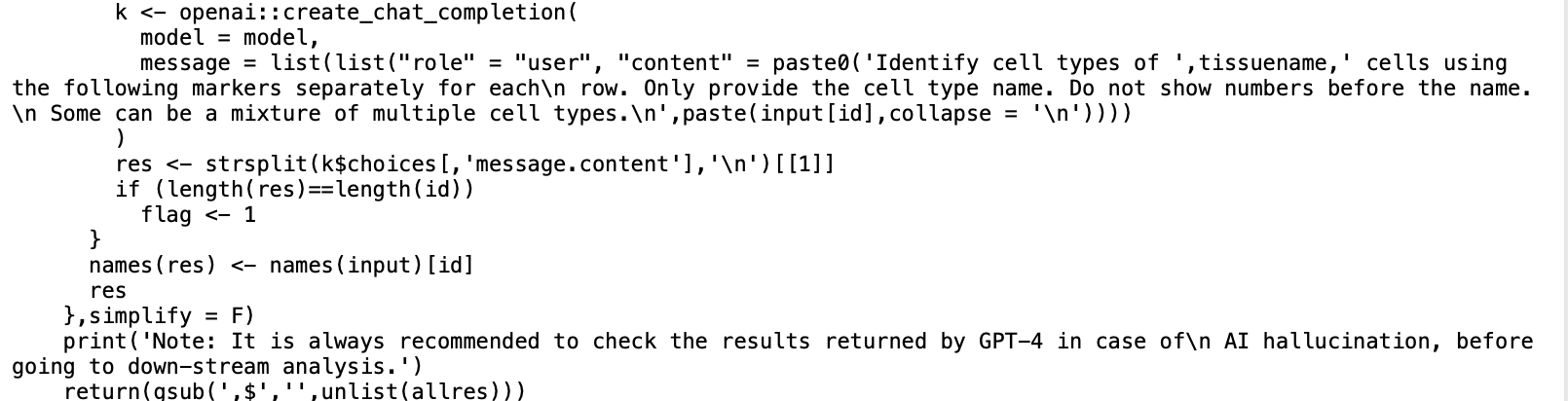
普通思路
如果你仔细看它这个思路最早是在预印本,Hou, W. and Ji, Z., 2023. Assessing GPT-4 for cell type annotation in single-cell RNA-seq analysis. bioRxiv, pp.2023-04, doi: https://doi.org/10.1101/2023.04.16.537094.,也就是说是 2023.04.16 ,但是我朋友圈的小伙伴早一个多月就测试了同样的思路:
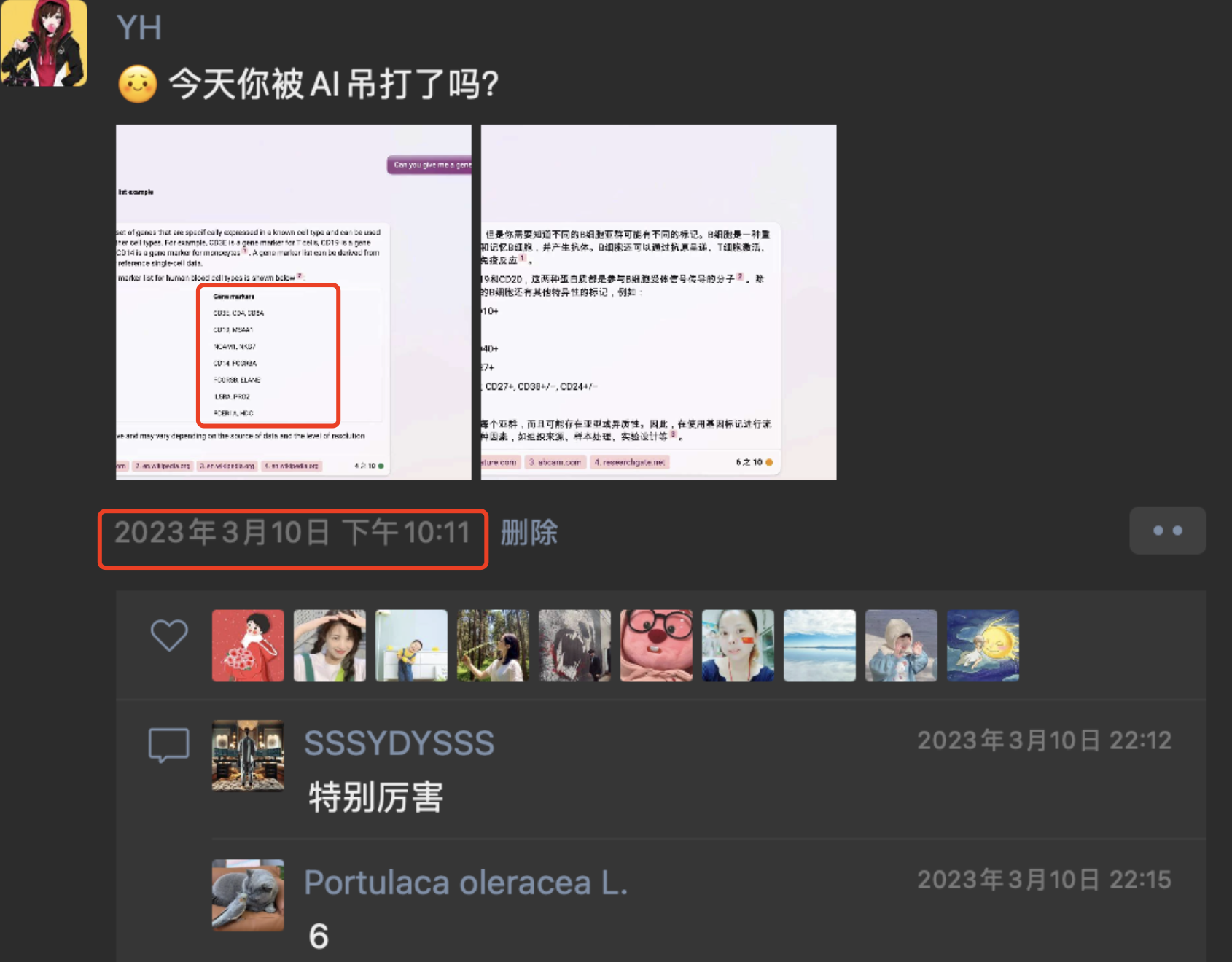
也就是说,思路本身并不高级,但是人家整理成为了文章而且丢预印本啦,而且最终成功发表在了 《Nature Methods》杂志,而我们只能说是发一个朋友圈或者一个公众号推文。
因为这个单细胞亚群的注释是刚需,常规的单细胞转录组降维聚类分群代码可以看 链接: https://pan.baidu.com/s/1bIBG9RciAzDhkTKKA7hEfQ?pwd=y4eh ,基本上大家只需要读入表达量矩阵文件到r里面就可以使用Seurat包做全部的流程,但是初始情况下只能说是拿到如下所示的图:
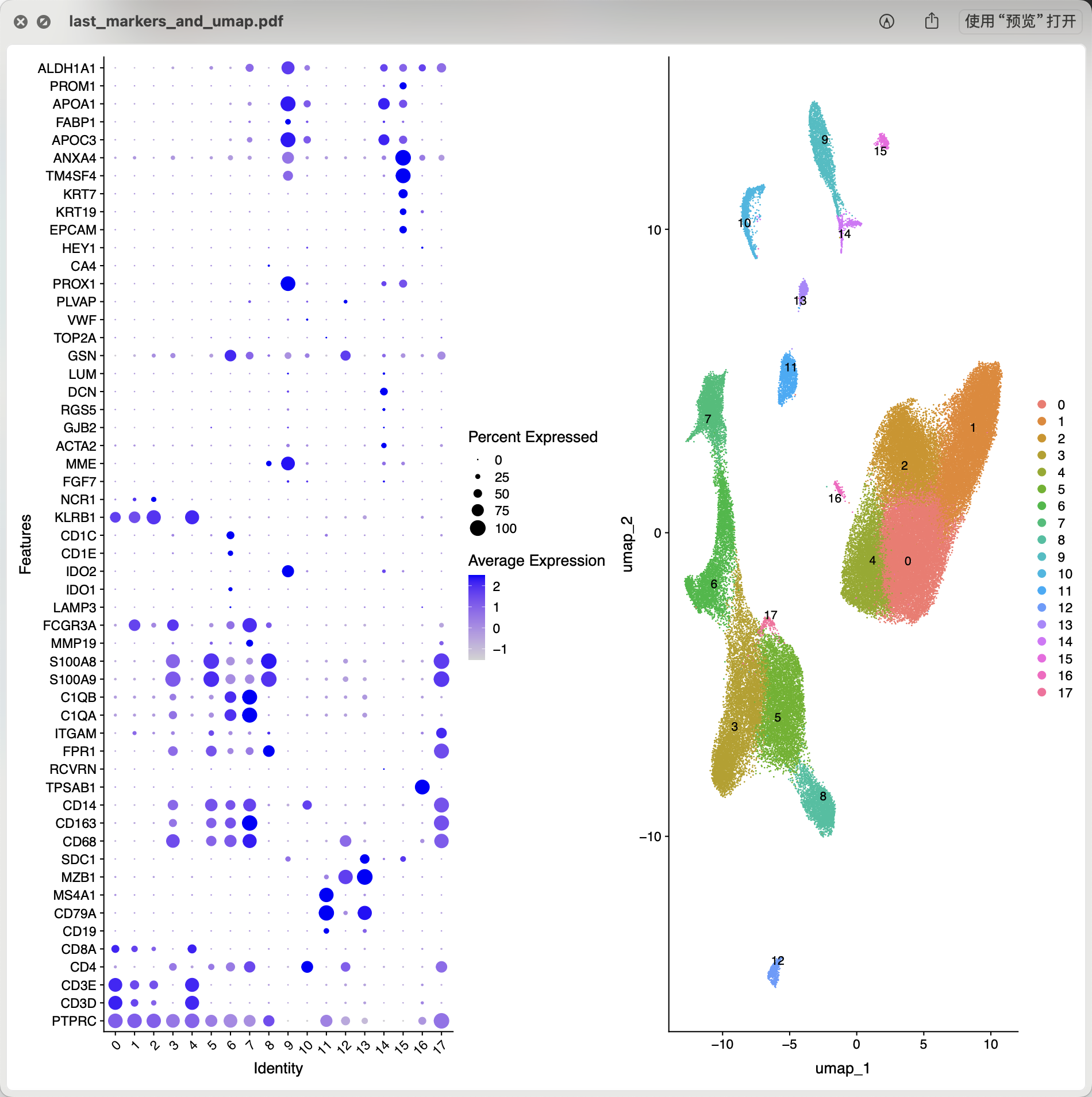
因为算法只能说是给出来顺序编号的亚群名字,仍然是需要大家自己背诵那些每个单细胞亚群的特异性基因后才能是给出来生物学命名。而且我们背诵的基因是有限的,有些时候部分亚群会漏掉,我们也可以找出来每个顺序编号的亚群的top基因,而不仅仅是是特异性基因,比如:
cluster0:CD3D,CD3G,CD3E,IL7R,IL32,LTB
cluster1:FGFBP2,GNLY,GZMB,GPR56,CX3CR1,SPON2
cluster2:XCL1,KLRC1,XCL2,IL2RB,CD160,TXK
cluster3:RP11-290F20.3,LILRB2,LYPD2,CDKN1C,LILRA5,FCN1
cluster4:TNF,DNAJB1,JUN,SLC4A10,HSPA1B,IL7R
cluster5:VCAN,S100A12,RP11-1143G9.4,EREG,THBS1,C19orf59
cluster6:CD1C,CLEC10A,CLEC9A,CD1E,FCER1A,IDO1
cluster7:CD5L,VCAM1,SDC3,FOLR2,MARCO,CXCL12
cluster8:FCGR3B,CMTM2,PTGS2,CXCR2,S100P,G0S2
cluster9:AFM,RP11-548L20.1,TENM2,SLC28A1,ACSM2B,ACSM2A
cluster10:PTPRB,STAB2,OIT3,ELTD1,CRHBP,AKAP12
cluster11:MS4A1,CD79A,LINC00926,BANK1,VPREB3,RP11-693J15.5
cluster12:LILRA4,MAP1A,SCT,LAMP5,IL3RA,LRRC26
cluster13:TNFRSF17,MZB1,RP11-731F5.2,RP11-16E12.2,DERL3,IGJ
cluster14:PTH1R,LAMA2,CCBE1,PDE1A,PRKG1,PDZRN4
cluster15:DCDC2,CHST4,SLC5A1,BICC1,CTNND2,KCNJ16
cluster16:TPSAB1,CPA3,CTSG,HPGDS,HDC,CMA1
cluster17:CTAG2,CYP1B1,FCAR,TMEM176A,SAP30,VSTM1
然后针对这些每个顺序编号的亚群的top基因去查询它们的单细胞亚群生物学名字!通常我们拿到了肿瘤相关的单细胞转录组的表达量矩阵后的第一层次降维聚类分群通常是:
- immune (CD45+,PTPRC),
- epithelial/cancer (EpCAM+,EPCAM),
- stromal (CD10+,MME,fibro or CD31+,PECAM1,endo)
参考我前面介绍过 CNS图表复现08—肿瘤单细胞数据第一次分群通用规则,这3大单细胞亚群构成了肿瘤免疫微环境的复杂。绝大部分文章都是抓住免疫细胞亚群进行细分,包括淋巴系(T,B,NK细胞)和髓系(单核,树突,巨噬,粒细胞)的两大类作为第二次细分亚群。但是也有不少文章是抓住stromal 里面的 fibro 和endo进行细分,并且编造生物学故事的。如下所示:

其实更麻烦的地方在于,我们第一层次降维聚类分群的时候往往是比较容易复现的:
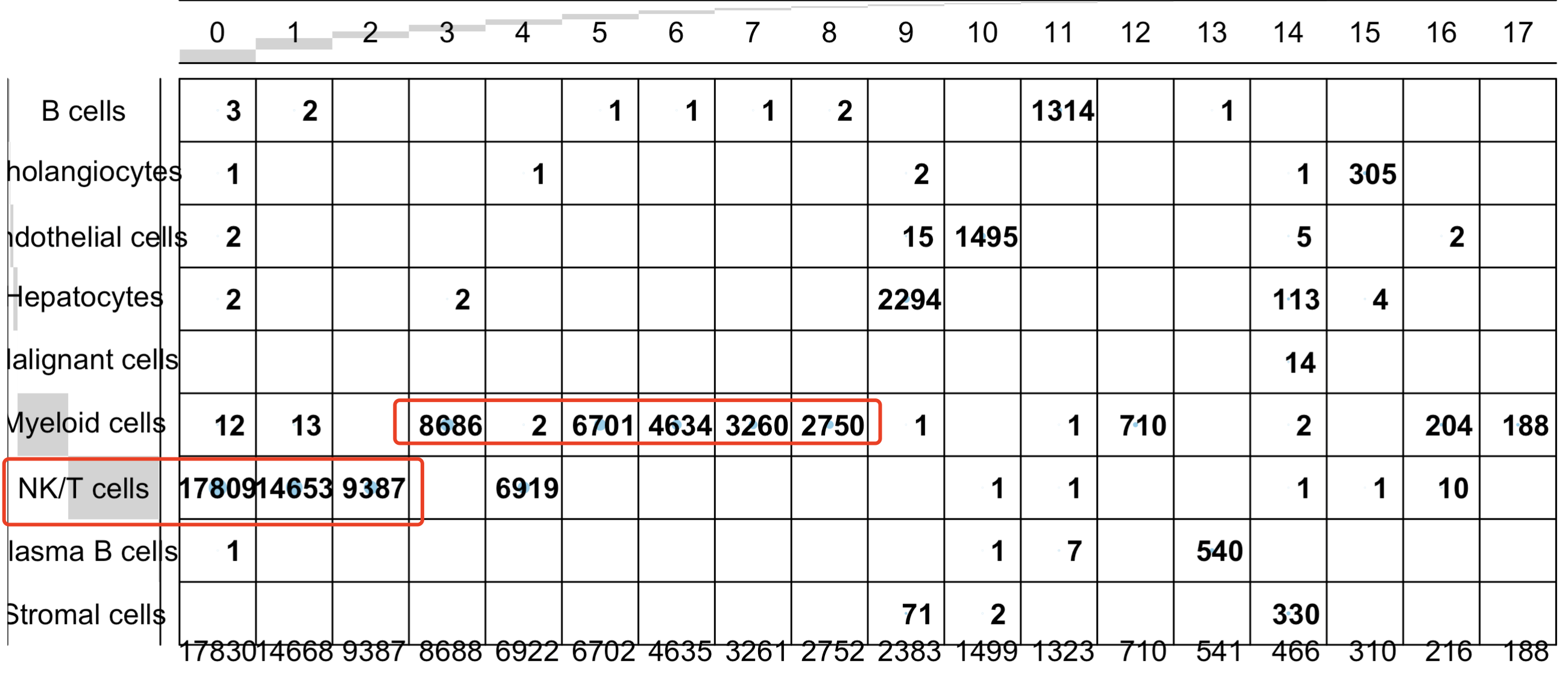
比如上面的顺序编号的0,1,2,4都是t或者nk细胞,但是它们在第一层次降维聚类分群的UMAP的二维坐标是很难有清晰界限的。也就是说 细分亚群的时候,其实是需要重新跑降维聚类分群了在每个子集细胞亚群里面:
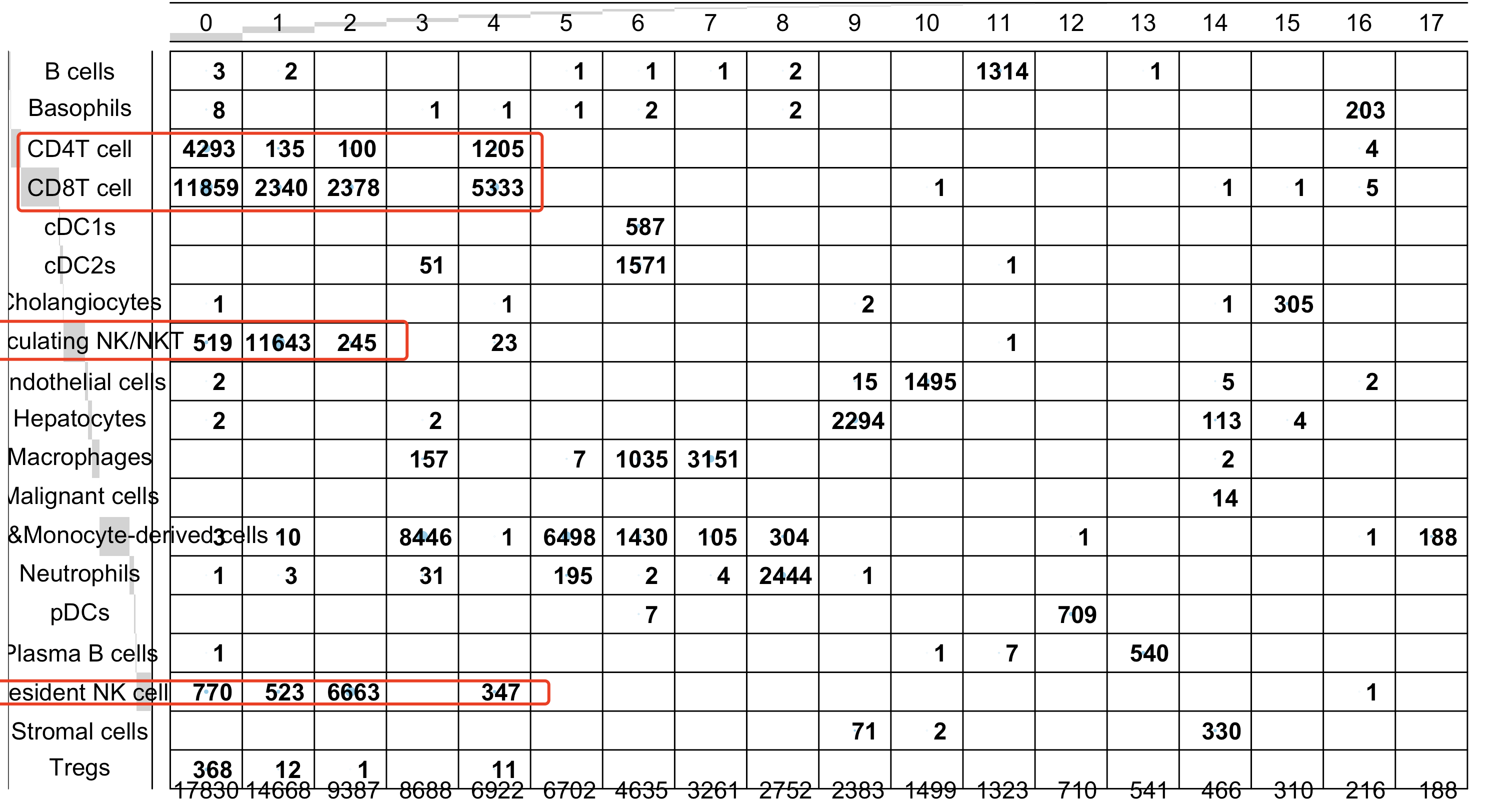
生物信息学期刊分级整理
主要是依据好久之前看到了知乎上面的一个生物信息学期刊整理,总结整理的很好好,感觉确实看过的大部分文献都在这8种杂志里面,如下所示:
主要是因为纯粹的生物信息学专业领域期刊的影响因子都不怎么高,所以大家才会去去综合性期刊投递生物信息学文章,传统生物信息学期刊主要是:
- Bioinformatics
- Briefings in Bioinformatics
- PLOS Computational Biology
- BMC Bioinformatics
- genome biology
如果是普通的生物信息学数据挖掘文章,就只能说是在3大出版社那边了,Frontiers 旗下的 以及MDPI旗下的