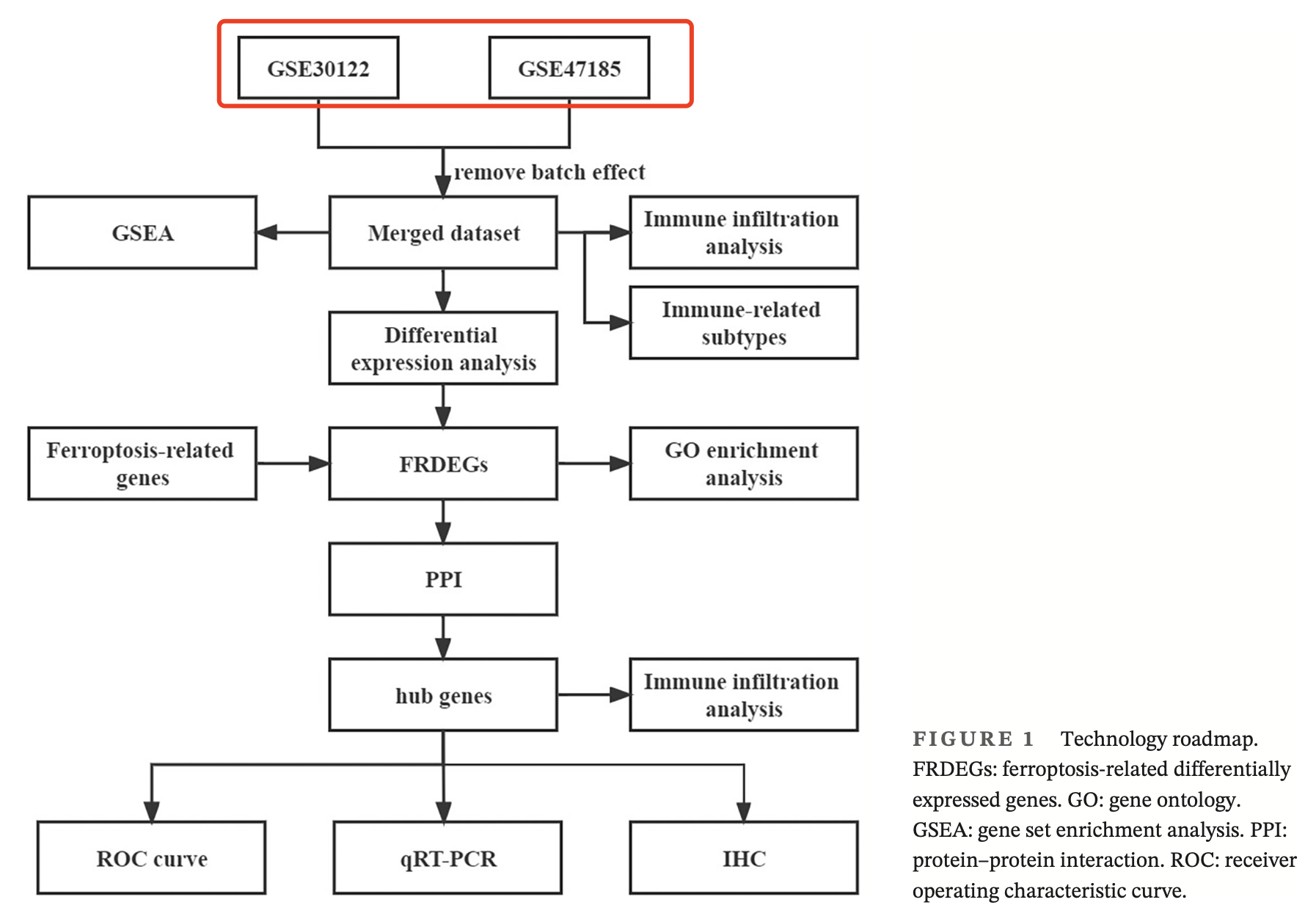
看到了一个生物信息学数据挖掘,标题是:《Novel ferroptosis gene biomarkers and immune infiltration profiles in diabetic kidney disease via bioinformatics》,开头就是两个表达量芯片数据集的去批次效应后的整合 :
但是可以看到其中一个芯片是有问题的,因为它的表达量在0附近,也就是说 它被zscore了,如下所示:
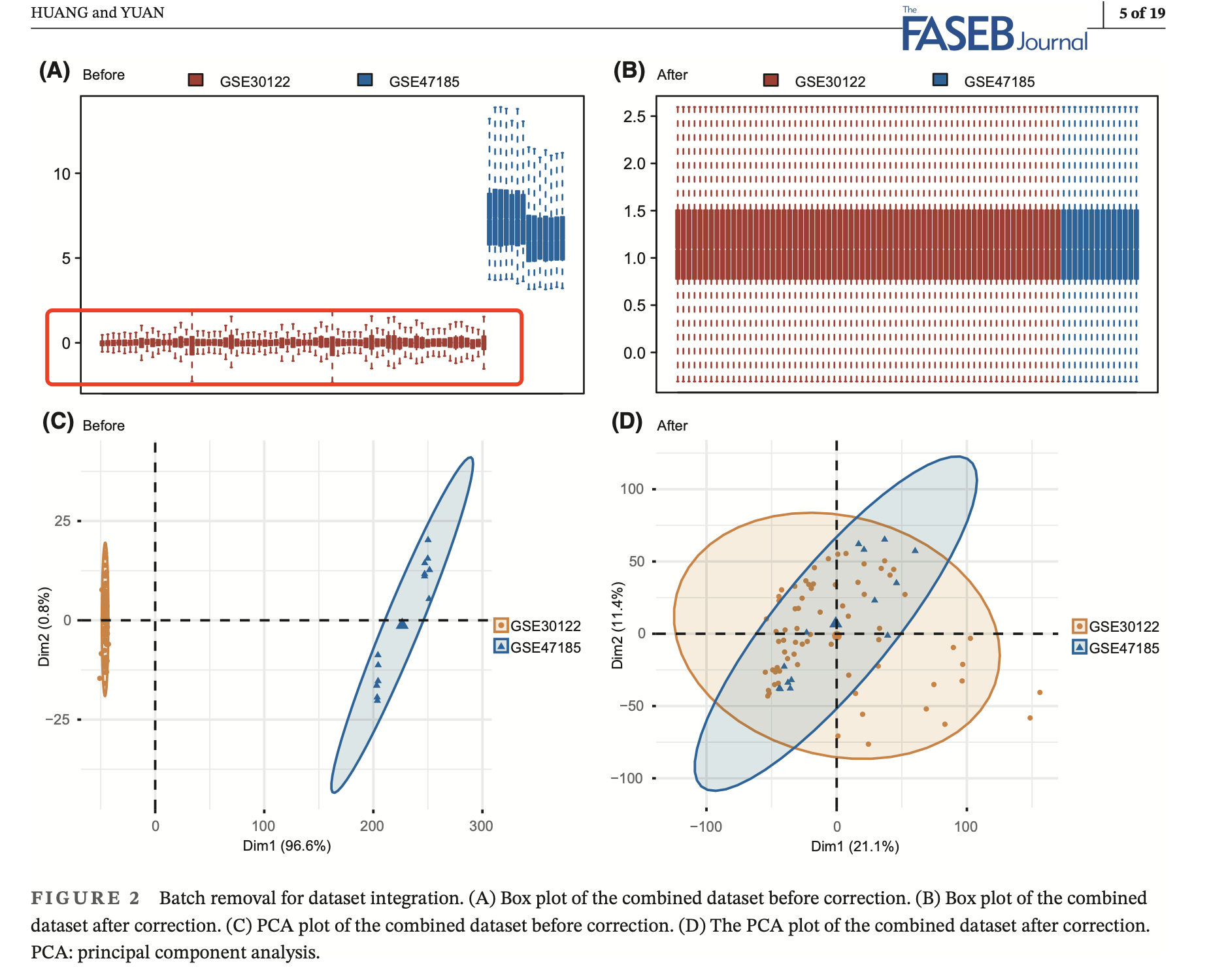
实际上我们不能如此简单粗暴的使用它这样的被zscore了表达量矩阵去做后续分析,这样去除表达量芯片的批次效应可能不妥,我们可以针对这个数据集去下载表达量芯片的原始数据来处理它。
正确的做法应该是?
我们可以去看看官网:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE30122
可以看到这个表达量芯片是非常经典的 Affymetrix Human Genome U133A 2.0 Array
每个样品都有cel文件的,可以读取它,自己制作表达量矩阵 :
Supplementary file Size Download File type/resource
GSE30122_RAW.tar 142.8 Mb (http)(custom) TAR (of CEL)
Custom GSE30122_RAW.tar archive:
Supplementary file File size
GSM756992_KS1-HG_U133A_2-2757.CEL.gz 2.1 Mb
GSM756993_KS1-HG_U133A_2-2871.CEL.gz 1.9 Mb
GSM756994_KS1-HG_U133A_2-2901.CEL.gz 2.0 Mb
GSM756995_KS1-HG_U133A_2-2905.CEL.gz 2.0 Mb
GSM756996_KS1-HG_U133A_2-5117.CEL.gz 2.0 Mb
之前我们也给出来了标准的代码:
rm(list = ls())
library(oligo)
library(ggplot2)
celFiles <- list.celfiles('GSE66676_RAW/',listGzipped = T,
full.name=TRUE)
celFiles
exon_data <- oligo::read.celfiles( celFiles )
dim(exprs(exon_data))
exprs(exon_data)[1:4,1:4]
### 标准化(一步完成背景校正、分位数校正标准化和RMA (robust multichip average) 表达整合)
exon_data_rma <- oligo::rma(exon_data )
exp_probe <- Biobase::exprs(exon_data_rma)
exp_probe[1:4,1:4]
我们会在这周六( 2024-05-25 )下午四点半直播分享这个正确的做法,以及这两种不同的的数据处理方案最后的数据分析结果的区别!
然后你会神奇的发现, 哪怕是文章作者使用了错误的数据处理方案,其实也并不会对它文章的结论造成很大的影响!!!