转录组测序定量流程大家都很熟悉了,一般来说我们推荐经典的4步:
-
1.数据准备(找公司测序的话,或者下载公共数据集)
-
2.质量控制(质控前用fastqc与multiqc初看数据效果、trim-galore进行质控过滤 )
-
3.使用Hisat2比对
-
4.使用featureCounts定量
很容易就拿到了count矩阵,但是早期大家喜欢RPKM(Reads Per Kilobase per Million reads)、FPKM(Fragments Per Kilobase of transcript per Million fragments)和TPM(Transcripts Per Million),这三种常用标准化指标。
目前我们还是就纯粹的count矩阵即可,如果大家的count矩阵来源于多个数据集,理论上就需要去批次啦。
首先我们有4个不同数据集的表达量矩阵
需要进行如下所示的简单的合并即可:
# 数据框 dat1, dat2, dat3, dat4 行名取交集
common_rows <- intersect(rownames(dat1),
intersect(rownames(dat2),
intersect(rownames(dat3),
rownames(dat4))))
# 使用 cbind 合并数据框
exp <- cbind(
dat1[common_rows, ],
dat2[common_rows, ],
dat3[common_rows, ],
dat4[common_rows, ]
)
Group = c(group_list1,group_list2,group_list3,group_list4)
table(Group)
getwd()
save(dat1,dat2,dat3,dat4,common_rows,Group,exp,file = "Rdata/exp.Rdata")
然后使用sva包的ComBat_seq函数针对转录组测序的count矩阵去批次
如下所示的代码:
rm(list = ls())
load("Rdata/exp.Rdata")
#处理批次效应(combat)
library(sva)
#ComBat_seq是基于ComBat框架的改进模型,专门针对 RNA-Seq 计数(即count矩阵)数据
batch <- c(rep("A", ncol(dat1)), rep("B", ncol(dat2)),
rep("C", ncol(dat3)), rep("D", ncol(dat4)))
mod = model.matrix(~Group)
exp2 = ComBat_seq(counts = as.matrix(exp), batch = batch,group = Group)
class(exp2)
dat = log2(edgeR::cpm(exp2)+1)
save(exp,exp2,dat,Group,file = "Rdata/dat.Rdata")
去除前后的表达量矩阵可以简单的看看主成分分析结果,如下所示,可以看到不同数据集的差异被抹除了 :
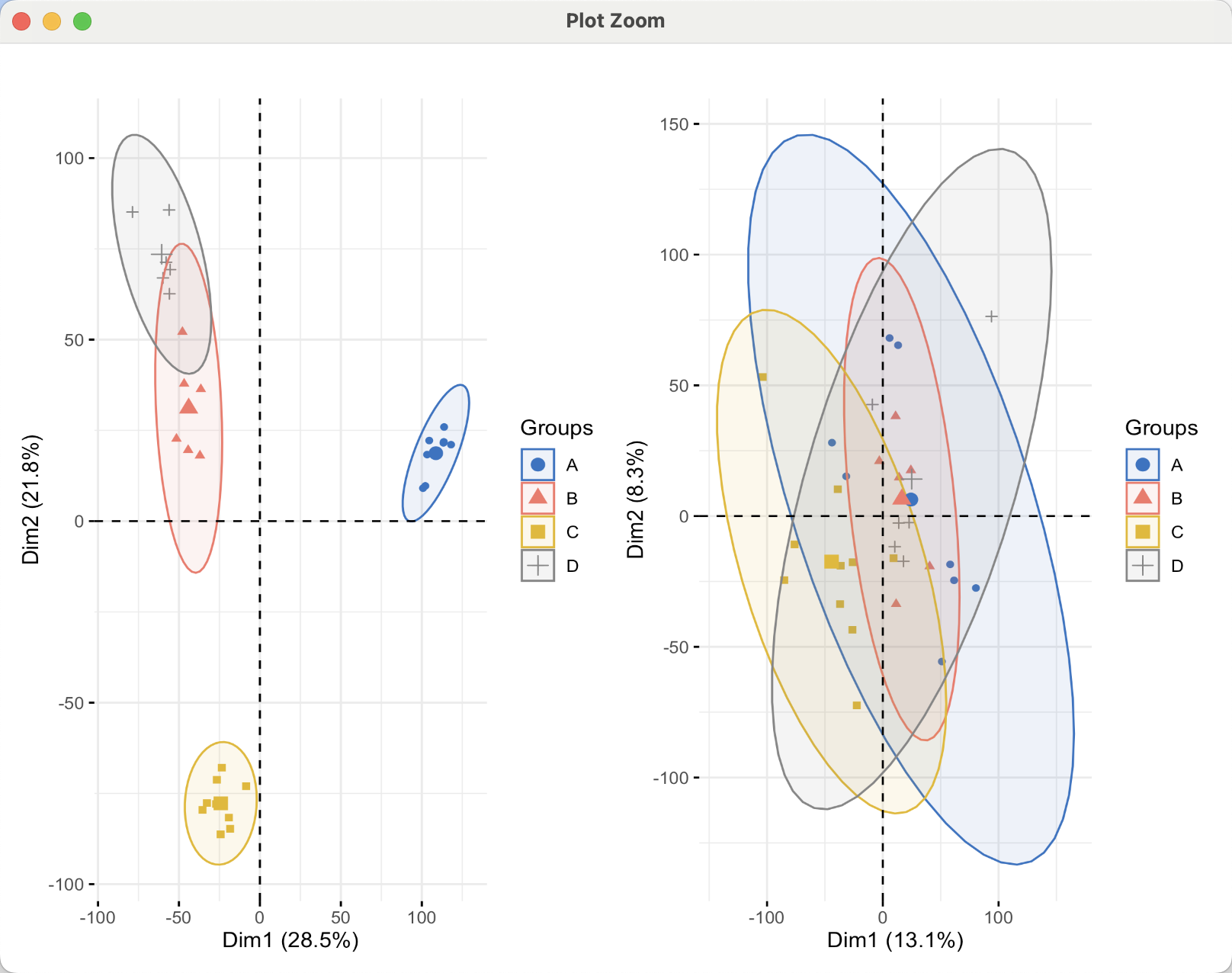
而且 它去除前后的表达量矩阵,都是count格式:
> exp[1:4,1:4]
GC_B13 GC_B14 GC_B2 GC_B30
TSPAN6 1276 1022 1417 1150
DPM1 798 767 1269 1023
SCYL3 91 108 38 50
C1orf112 75 86 71 33
> exp2[1:4,1:4]
GC_B13 GC_B14 GC_B2 GC_B30
TSPAN6 392 204 702 405
DPM1 294 283 470 379
SCYL3 97 108 49 59
C1orf112 46 57 43 15
是可以走后面的常规的转录组差异分析流程的!