前些天在朋友圈看到了小伙伴分享了张泽民老师的一个最新单细胞文章:《Spatiotemporal single-cell analysis decodes cellular dynamics underlying different responses to immunotherapy in colorectal cancer》,数据集是GSE236581,是 primary tumor tissues, adjacent normal tissues, and peripheral blood of 22 CRC patients underwent neoadjuvant anti-PD-1 treatment. 总计 169 single-cell samples ,数据量确实是非常可观。
小伙伴表示如果是在r编程语言里面处理它, 仅仅是读取就耗费25分钟啦。我们可以在其GEO界面(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE236581) 看到研究者们公开的,如下所示的文件:
GSE236581_CRC-ICB_metadata.txt.gz 12.7 Mb
GSE236581_VDJ_merge.txt.gz 43.0 Mb
GSE236581_barcodes.tsv.gz 4.5 Mb
GSE236581_counts.mtx.gz 3.9 Gb
GSE236581_features.tsv.gz 243.5 Kb
下载这些文件后简单的修改名字和文件夹结构,如下所示:
tree -h inputs/
[ 160] inputs/
├── [4.5M] barcodes.tsv.gz
├── [244K] features.tsv.gz
└── [3.9G] matrix.mtx.gz
读取本身是很简单的事情(就是好费时间,而且取决于计算机资源):
ct=Read10X('inputs/',gene.column = 1)
dim(ct)
#[1] 36027 975275
可以看到是接近100万个细胞啦,而且研究者们给出来了比较好的单细胞亚群注释信息:
phe=data.table::fread('GSE236581_CRC-ICB_metadata.txt.gz',data.table = F)
head(phe)
rownames(phe)=phe[,1]
phe=phe[,-1]
table(phe$Ident)
gplots::balloonplot(
table(phe$MajorCellType,phe$Tissue)
)
如下所示:
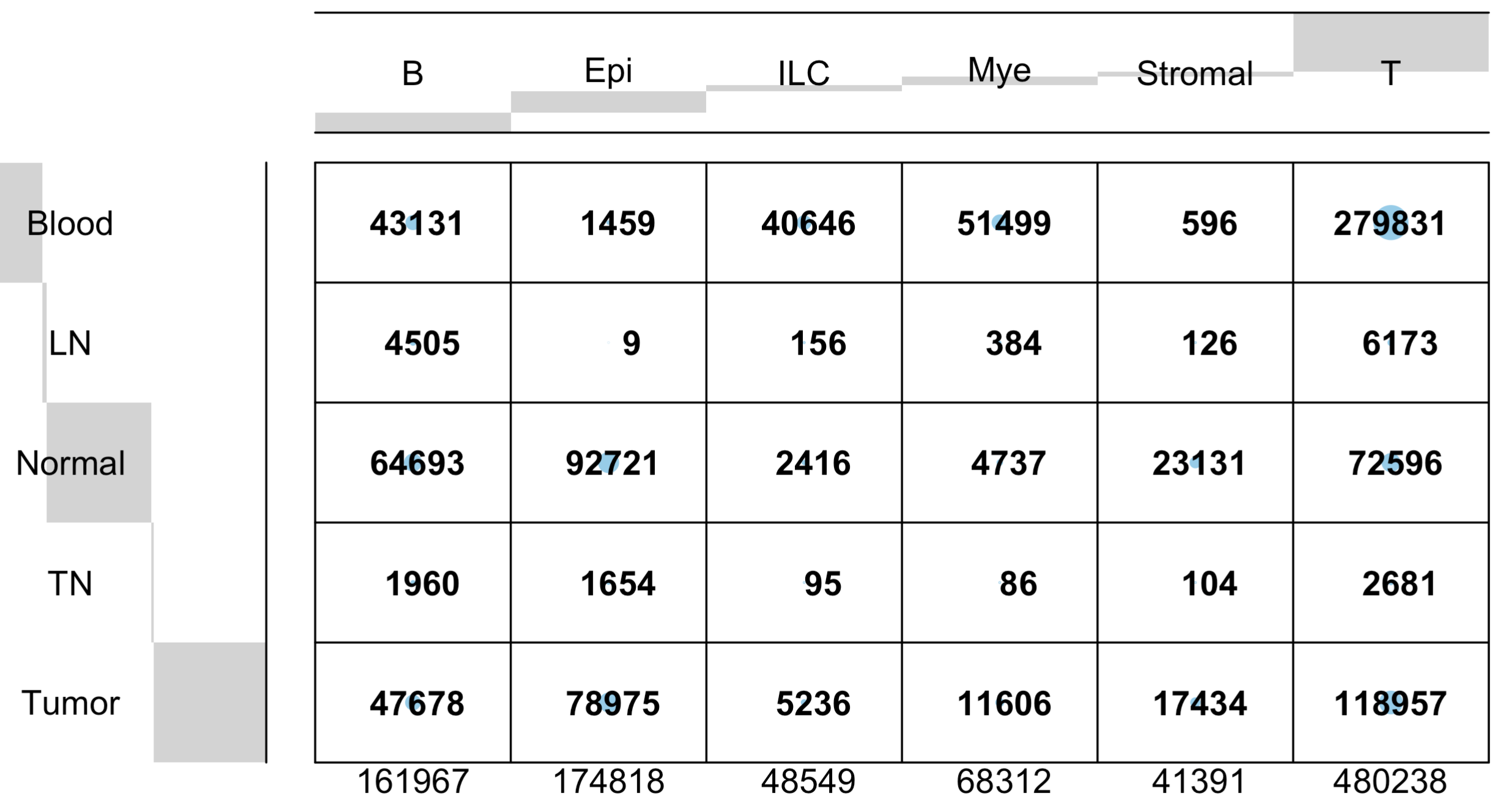
也就是说,大家在重新分析这个数据集的时候,其实已经是不需要重新对这么大一个数据集进行降维聚类分群啦。起码第一层次降维聚类分群和第二层次作者都给出来了,而且很清晰:
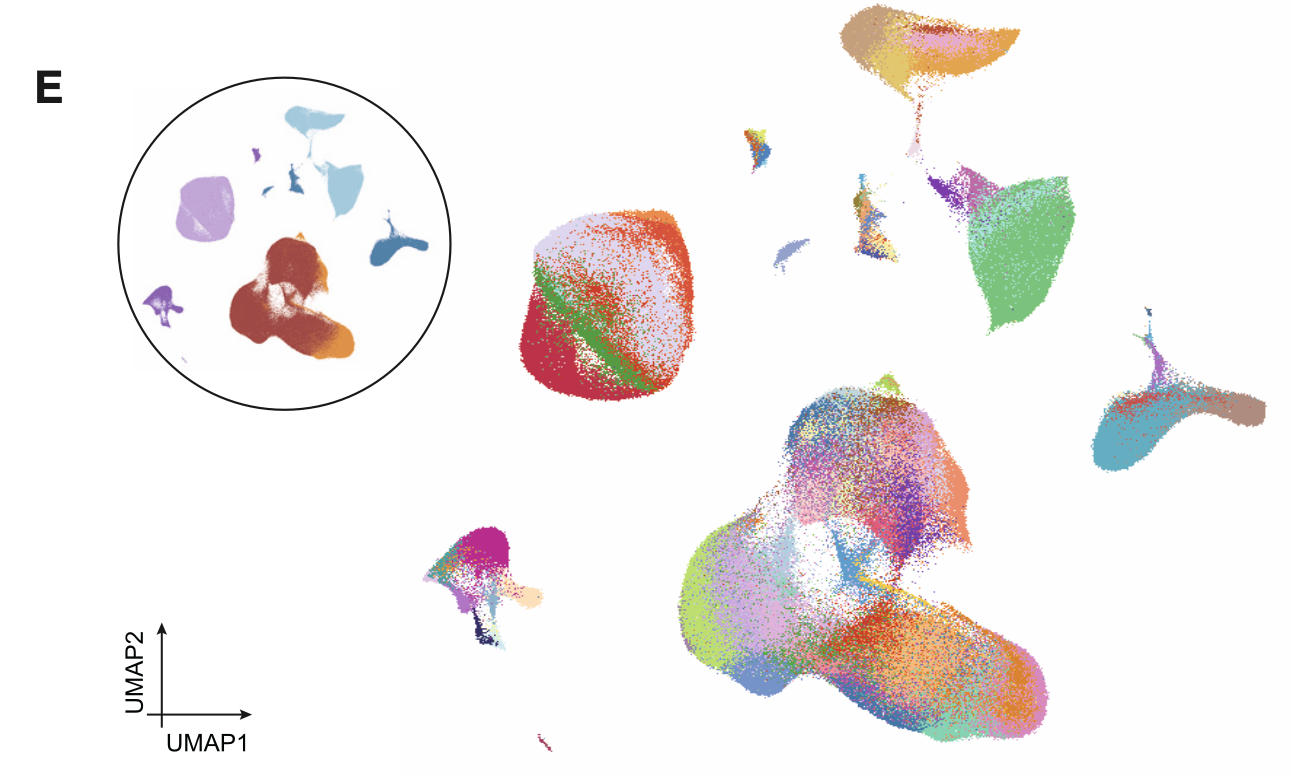
也就是说,对GSE236581这个数据集来说,读取它全部的接近100万个细胞后走单细胞转录组流程其实是伪命题啦,不过,也确实是很多公共数据集并不会给作者注释好的信息,或者说有时候作者自己的注释也并不完善或者让大家信服, 就需要自己从零开始处理啦。
如果你是一定要亲自拿到上面的降维聚类分群后的UMAP图,在r里面就只能说是借助配置超级好的计算机。如果你也想做单细胞转录组数据分析,最好是有自己的计算机资源哦,比如我们的2024的共享服务器交个朋友福利价仍然是800,而且还需要有基本的生物信息学基础,也可以看看我们的生物信息学马拉松授课(买一得五) ,你的生物信息学入门课。
值得注意的是,八月份比较特殊的是:单细胞数据挖掘线下培训(南京站),下周六日我们可以线下授课,惊喜不!