有粉丝在交流群提问说看到了2022的一个泛癌文章但是数据量有点大以至于他自己的个人电脑或者课题组服务器都没办法hold住,标题是 :《Pan-cancer single-cell analysis reveals the heterogeneity and plasticity of cancer- associated fibroblasts in the tumor microenvironment》,这个泛癌单细胞数据挖掘文章纳入了很多不同癌症的单细胞转录组数据集做了一个汇总的降维聚类分群,如下所示:

因为纳入的数据集有点多,来源于12篇文章:232 single cell transcriptome samples (normal = 31; adjacent = 54; tumor = 148) ,分别来源于:
- 31 GSM samples were derived from GSE134355
- 11 GSM samples wererelated to GSE141445
- 49 samples were derived from the under accession numberE-MTAB-8107
- 2 GSM samples were associated with GSE157703
- 22 GSMsamples were derived from GSE131907
- 7 GSM samples were derived fromGSE138709
- 31 samples were derived from the under accession number E-MTAB-6149 and E-MTAB-6653
- 32 CRR samples were related to GSA database underaccession number CRA001160
- 10 GSM samples were related to GSE154778
- 7 HRR samples were available in GSA-Human under the accession codeHRA000212
- 10 thyroid samples were derived from GSA-Human databaseunder accession number HRA000686
- 20 gastric samples were download from http://dna-discovery.stanford.edu/download/1401/;
并不需要自己去重新分析上面的12个文章的12个数据集了,因为作者直接就在 GSE210347 数据集给出来了表达量矩阵文件 (GSE210347_counts.Rds.gz ),如下所示:
GSE210347_counts.Rds.gz 2.4 Gb (ftp)(http) RDS
GSE210347_meta.txt.gz 18.5 Mb (ftp)(http) TXT
GSE210347_sample_information.txt.gz 632 b (ftp)(http) TXT
GSE210347_study_metadata.xls.gz 39.8 Kb (ftp)(http) XLS
其实这个甚至都不需要自己做降维聚类分群了,因为本来就是有对应的GSE210347_meta.txt.gz 18.5 Mb 文件,里面就给每个细胞一个身份了。如果假设作者没有提供,我们就需要加载作者的表达量矩阵文件 (GSE210347_counts.Rds.gz )然后走降维聚类分群流程啦。
拆分这个表达量矩阵
我做了一个简单的 操作:
tmp = readRDS('../inputs/GSE210347_counts.Rds')
dim(tmp) # [1] 42113 855271
half_cols <- ceiling(ncol(tmp) / 2);half_cols
### part 1
sce.all=CreateSeuratObject(
counts = tmp[,1:half_cols]
)
### part 2
sce.all=CreateSeuratObject(
counts = tmp[,(half_cols+1):ncol(tmp)]
)
上面的 855271 个细胞,被我平等的拆分成为了两个矩阵后各自构建了 Seurat对象,只需要对 两个 sce.all 变量 走我们的降维聚类分群流程即可。唯一的区别就是文章里面的分群UMAP图就是一个:
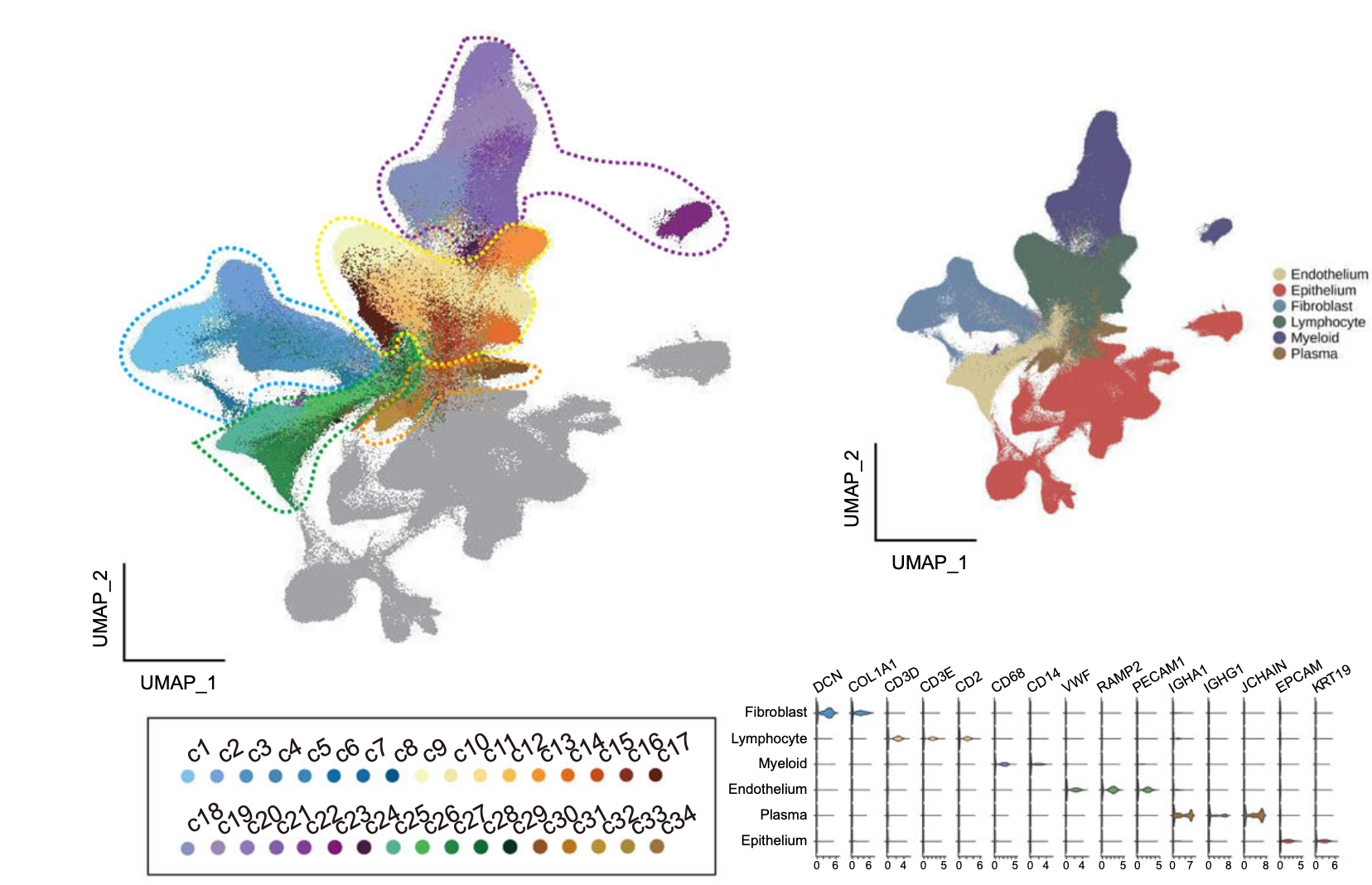
但是我们把表达量矩阵拆分了,所以就会跑出来多个分群UMAP图,不过这个并不重要,因为我们之所以要跑降维聚类分群流程就是为了给细胞一个身份,这个过程甚至是可以免去降维聚类分群流程的,因为有很多自动化注释软件,它们是针对具体的每个单细胞本身独立的注释。
比如,我们可以使用作者的降维聚类分群和细胞亚群命名结果来验证一下我们的拆分成为两个单细胞表达量矩阵之后的结果:
library(data.table)
tmp = fread('../inputs/GSE210347_meta.txt.gz',
data.table = F)
> colnames(tmp)
[1] "cellname" "nCount_RNA" "nFeature_RNA"
[4] "SampleID" "percent.mt" "seurat_clusters"
[7] "cluster" "celltype" "tissue"
[10] "group"
> table(tmp$celltype)
Endothelium Epithelium Erythrocyte Fibroblast Lymphocyte Myeloid
74102 268469 3295 104804 202077 151298
Plasma undefined
37638 13588
如下所示,哪怕是拆分了,结果仍然是还不错:
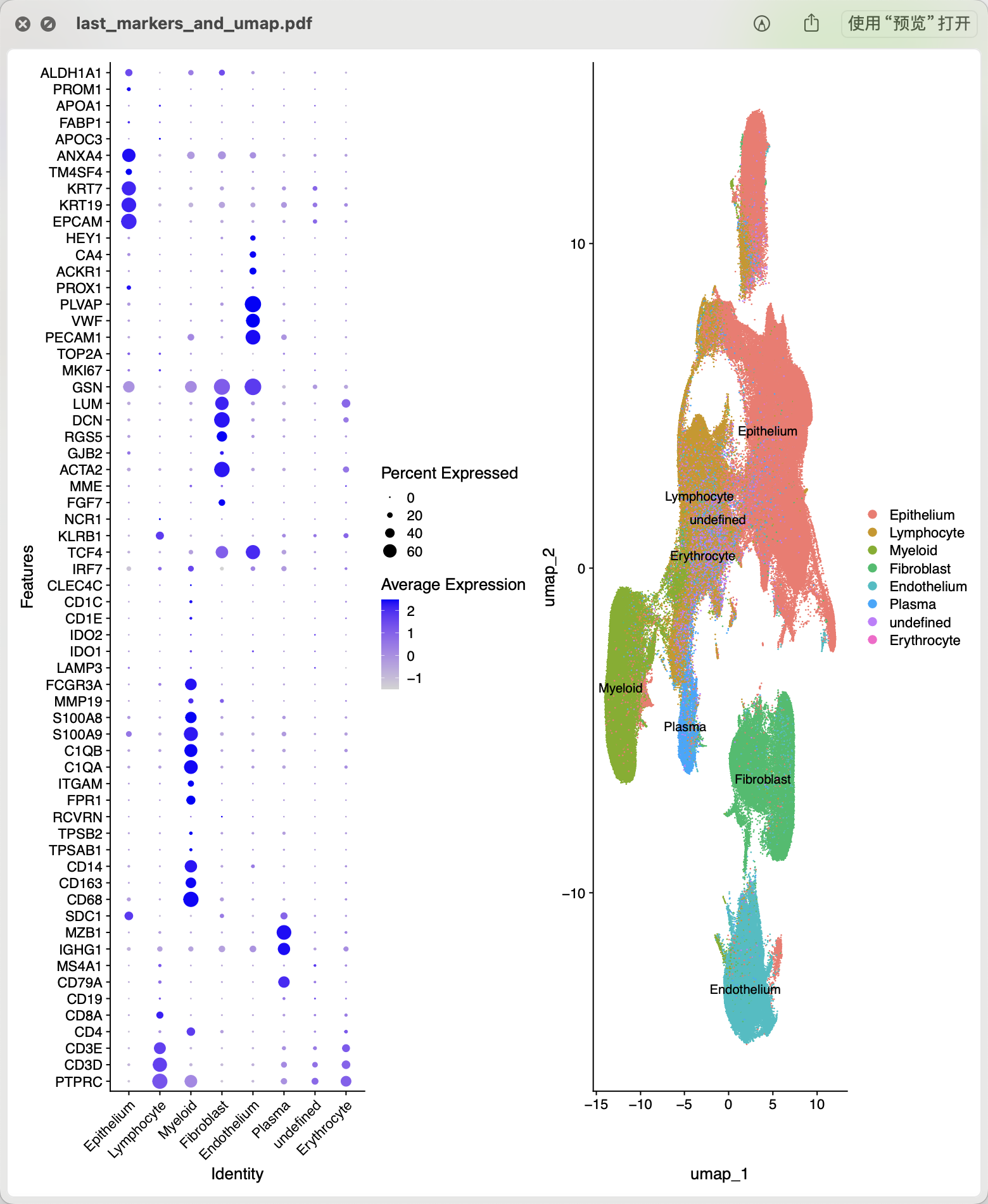
而且绝大部分小伙伴拿到了主要的百万级别单细胞转录组数据集,其实并不会关心全局情况,应该是会挑选里面的具体的某个单细胞亚群,比如癌症相关成纤维细胞,然后对它继续细致的降维聚类分群后讨论它的临床意义。
当然了,如果你想完成上面的操作,最好是有自己的计算机资源哦,比如我们的2024的共享服务器交个朋友福利价仍然是800,而且还需要有基本的生物信息学基础,也可以看看我们的生物信息学马拉松授课(买一得五) ,你的生物信息学入门课。
如果你有了生物信息学基础,仅仅是感兴趣单细胞的细致分析,就看看SBC的 Nature年度技术 | 单细胞及空间多组学实验技术与生信分析培训(暑期班)招生火热进行中