单细胞转录组数据分析最基础的就是给每个细胞一个身份,通常是降维聚类分群后然后对每个亚群进行描述,首先可以描述每个亚群的高表达量的特异性基因,然后可以对基因进行生物学功能数据库注释。也可以直接对亚群进行打分,比如gsea和gsva算法,针对免疫或者代谢等基因集进行打分,也是拿到每个亚群的特异性生物学功能。
之前我们介绍过irGSEA:基于秩次的单细胞基因集富集分析整合框架,针对17种常见的Functional Class Scoring (FCS)方法进行了benchmark,感兴趣的可以仔细读一下。最近恰好看到了密西根大学的Research Assistant Professor Neurology的Kai Guo的github也有一个打分工具:https://github.com/guokai8/scGSVA ,也值得介绍一下:
基于基因列表的生物学功能数据库注释
单细胞转录组降维聚类分群大家都应该是很熟悉了,有了分群结果就可以很容易找到每个亚群的特异性基因列表,然后基于每个亚群的基因列表就可以进行生物学功能数据库注释,代码如下所示:
###热图表示GO富集分析
library(grid)
library(ggplot2)
library(gridExtra)
library(clusterProfiler)
library(org.Hs.eg.db)
library(enrichplot)
library(ggplot2)
library(org.Hs.eg.db)
library(GOplot)
if (!file.exists('markers.csv')) {
markers <- FindAllMarkers(object = sce, only.pos = TRUE,
min.pct = 0.25,
thresh.use = 0.25)
write.csv(markers,file='markers.csv')
} else {
markers = read.csv('markers.csv',row.names = 1)
}
2. 如下所示的差异基因结果
table(markers$cluster)
#>
#> AT1 AT2 Cancer(CRABP2+) Cancer(TM4SF1+)
#> 1207 895 586 864
#> Cancer(UBE2C+) Ciliated cells Clara cells Clara-like cancer
#> 2278 676 2687 943
library(dplyr)
gene = markers[markers$p_val <0.01 & markers$avg_log2FC >2,]$gene
entrezIDs = bitr(gene, fromType = "SYMBOL", toType = "ENTREZID", OrgDb= "org.Hs.eg.db", drop = TRUE)
#> Warning in bitr(gene, fromType = "SYMBOL", toType = "ENTREZID", OrgDb =
#> "org.Hs.eg.db", : 7.1% of input gene IDs are fail to map...
gene<- entrezIDs$ENTREZID
marker_new = markers[markers$gene %in% entrezIDs$SYMBOL,]
marker_new = marker_new[!duplicated(marker_new$gene),]
identical(marker_new$gene, entrezIDs$SYMBOL)
#> [1] FALSE
p = identical(marker_new$gene, entrezIDs$SYMBOL);p
#> [1] FALSE
if(!p) entrezIDs = entrezIDs[match(marker_new$gene,entrezIDs$SYMBOL),]
marker_new$ID = entrezIDs$ENTREZID
3. 细胞亚群GO富集分析(基于compareCluster函数)
## GO
gcSample=split(marker_new$ID, marker_new$cluster)
###参数可以更改,看看?compareCluster
#One of "groupGO", "enrichGO", "enrichKEGG", "enrichDO" or "enrichPathway"
xx <- compareCluster(gcSample,
fun = "enrichGO",
OrgDb = "org.Hs.eg.db",
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 0.01,
qvalueCutoff = 0.05
)
p <- dotplot(xx)
library(ggthemes)
p + theme(axis.text.x = element_text(
angle = 45,
vjust = 0.5, hjust = 0.5
))+theme_few()
可以看到,每个单细胞亚群的生物学功能是有特异性的:

基于单细胞表达量矩阵的gsea和gsva(需要生物学功能数据库)
很容易从Seurat对象里面的拿到了单细胞表达量矩阵,一般来说都是两三万个基因然后几万个甚至几十万细胞数量,所以不推荐这个方法学,因为计算资源消耗比较大而且很慢,而且单细胞表达量矩阵里面的drop-out非常严重,这个时候Functional Class Scoring (FCS)方法并不好用!
基于细胞亚型表达量矩阵的gsea和gsva(需要生物学功能数据库)
其实就是针对Seurat对象进行AverageExpression函数操作一下,之前的几万个甚至几十万细胞数量就变成了几个或者十几个细胞亚群啦。因为每个细胞内部的成百上千个细胞被汇总了,所以基本上就不存在drop-out现象了,代码如下所示:
###GSVA分析
library(Seurat)
library(msigdbr)
library(GSVA)
library(tidyverse)
library(clusterProfiler)
library(patchwork)
library(limma)
#genesets <- msigdbr(species = "Homo sapiens")
genesets <- msigdbr(species = "Homo sapiens", category = "C2")
genesets <- subset(genesets, select = c("gs_name","gene_symbol")) %>% as.data.frame()
genesets <- split(genesets$gene_symbol, genesets$gs_name)
Idents(sce) <- sce$celltype
expr <- AverageExpression(sce, assays = "RNA", slot = "data")[[1]]
expr <- expr[rowSums(expr)>0,] #选取非零基因
expr <- as.matrix(expr)
head(expr)
#> Clara cells AT2 Cancer(UBE2C+) Clara-like cancer
#> AL627309.1 0.001611424 0.000000000 0.0012074311 0.0007027198
#> AL669831.5 0.083969542 0.049562047 0.0552978566 0.0595487007
#> FAM87B 0.000000000 0.000000000 0.0003532046 0.0000000000
#> LINC00115 0.012537201 0.023884735 0.0211631772 0.0284589549
#> FAM41C 0.091875324 0.032304186 0.0916539462 0.0353727547
#> AL645608.3 0.000000000 0.002181691 0.0050477681 0.0000000000
# gsva默认开启全部线程计算
gsva.res <- gsva(expr, genesets, method="gsva")
#> Warning: Calling gsva(expr=., gset.idx.list=., method=., ...) is deprecated;
#> use a method-specific parameter object (see '?gsva').
#> Warning in .gsva(expr, mapped.gset.idx.list, method, kcdf, rnaseq, abs.ranking,
#> : Some gene sets have size one. Consider setting 'min.sz > 1'.
#> Estimating GSVA scores for 6365 gene sets.
#> Estimating ECDFs with Gaussian kernels
# 。。。。。
gsva.df <- data.frame(Genesets=rownames(gsva.res), gsva.res, check.names = F)
gsva_d = gsva.res[sample(nrow(gsva.res),30),]
pheatmap::pheatmap(gsva_d, show_colnames = T,
scale = "row",angle_col = "45",
color = colorRampPalette(c("navy", "white", "firebrick3"))(50))
上面的gsva打分矩阵之后可以 pheatmap::pheatmap 的热图展示,也可以气泡图展现哈。
关于Kai Guo的scGSVA
我们可以首先借助ai了解一下,在生物信息学中,GSEA(Gene Set Enrichment Analysis)和GSVA(Gene Set Variation Analysis)是两种用于理解和解释基因表达数据的分析方法,它们都与基因集(通常是功能相关的一组基因)的分析有关。
GSEA(基因集富集分析)
GSEA 是一种计算方法,用于确定一个预先定义的基因集在某个生物状态下是否表现出显著的表达变化。这种分析方法基于排名,而不是统计测试,来评估基因集在不同样本或条件下的表达差异。GSEA的主要特点包括:
- 基因集:使用预先定义的基因集,如功能相关的基因、已知的通路成员或特定生物学过程的基因。
- 排名:基于基因表达的变化,对所有基因进行排名。
- 统计检验:通过计算基因集成员在排名列表中的位置,评估基因集在特定状态下是否富集在排名的顶端或底端。
- 多个测试:可以对多个基因集进行富集分析,以识别在特定条件下最受影响的生物学过程或通路。
- 可视化:通常以图表形式展示结果,如富集得分(ES)和名义P值或校正P值。
GSEA的主要用途是识别在疾病状态、发育阶段或对治疗响应中起作用的生物学通路。
GSVA(基因集变异分析)
GSVA 是一种用于评估基因集在不同样本或条件下变异的方法,它可以提供基因集水平上的表达变化信息,而不是单个基因。GSVA的主要特点包括:
- 表达矩阵:分析从RNA-seq或其他基因表达技术获得的表达矩阵。
- 变异度量:计算基因集的平均表达水平,并评估其在不同样本或条件下的变异。
- 平滑处理:使用平滑技术减少噪声,提高结果的可靠性。
- 统计分析:通过比较不同组之间的基因集表达差异,进行统计检验。
- 结果解释:识别在特定条件下表达变化显著的基因集,从而推断生物学过程或通路的变化。
GSVA的主要用途是识别在不同条件下基因集表达的变异性,这可能与疾病、发展或其他生物学变化有关。
区别和联系
- GSEA:侧重于基因集的富集分析,适用于探索特定生物学过程或通路在不同状态下的活性变化。
- GSVA:侧重于基因集表达的变异性分析,适用于评估基因集在不同条件下的整体表达变化。
两种方法都有助于从基因集的角度理解复杂的生物学数据,但它们在分析方法和应用场景上有所不同。GSEA更多用于识别特定状态下活跃的通路,而GSVA则用于评估基因集表达的变异性。在实际应用中,研究者可能会根据研究目的和数据类型选择合适的分析方法。
让我们直接实战吧:
#Installation
library(devtools)
install_github("guokai8/scGSVA")
#Examples
set.seed(123)
library(scGSVA)
data(pbmcs)
hsko<-buildAnnot(species="human",keytype="SYMBOL",anntype="KEGG")
res<-scgsva(pbmcs,hsko,method="ssgsea") ## or use UCell
vlnPlot(res,features="Wnt.signaling.pathway",group_by="groups") ## split.plot = TRUE and split.by
拆解一下上面的代码
是3个环节,构建数据库+打分+可视化。
其中构建数据库我前面使用的的是msigdbr这个包内置的 :
library(msigdbr)
#genesets <- msigdbr(species = "Homo sapiens")
genesets <- msigdbr(species = "Homo sapiens", category = "C2")
genesets <- subset(genesets, select = c("gs_name","gene_symbol")) %>% as.data.frame()
genesets <- split(genesets$gene_symbol, genesets$gs_name)
Kai Guo使用的是buildAnnot函数,可以打开代码看看,是基于KEGGREST keggLink在线获取的。
然后打分,首先是针对Seurat对象获取单细胞表达量矩阵
input <- GetAssayData(obj,assay = assay, layer = slot)
input<- input[tabulate(summary(input)$i) != 0, , drop = FALSE]
input <- as.matrix(input)
然后也是嫌弃gsva速度太慢了,所以拆分成为多个矩阵
split.data <- split.data.matrix(matrix=input, chunk.size=batch)
out<- lapply(split.data,function(x).sgsva(input=x,annotation = annotation,
method=method,kcdf=kcdf,
abs.ranking=abs.ranking,
min.sz=min.sz,
max.sz=max.sz,cores=cores,
tau=tau,ssgsea.norm=ssgsea.norm,
verbose=verbose
))
out <- do.call(rbind,out)
当然了,也可以使用其它打分方法啦:
out<- suppressWarnings(UCell::ScoreSignatures_UCell(input, features=annotation,
chunk.size = batch, ncores = cores,
BPPARAM=SerialParam(progressbar=verbose),...))
colnames(out)<-gsub(' ','\\.',sub('_UCell','',colnames(out)))
out<-as.data.frame(out)
最后是可视化,基本上就是Seurat包的5种可视化,打开源代码可以看到是重新使用ggplot2语法绘制了一下:
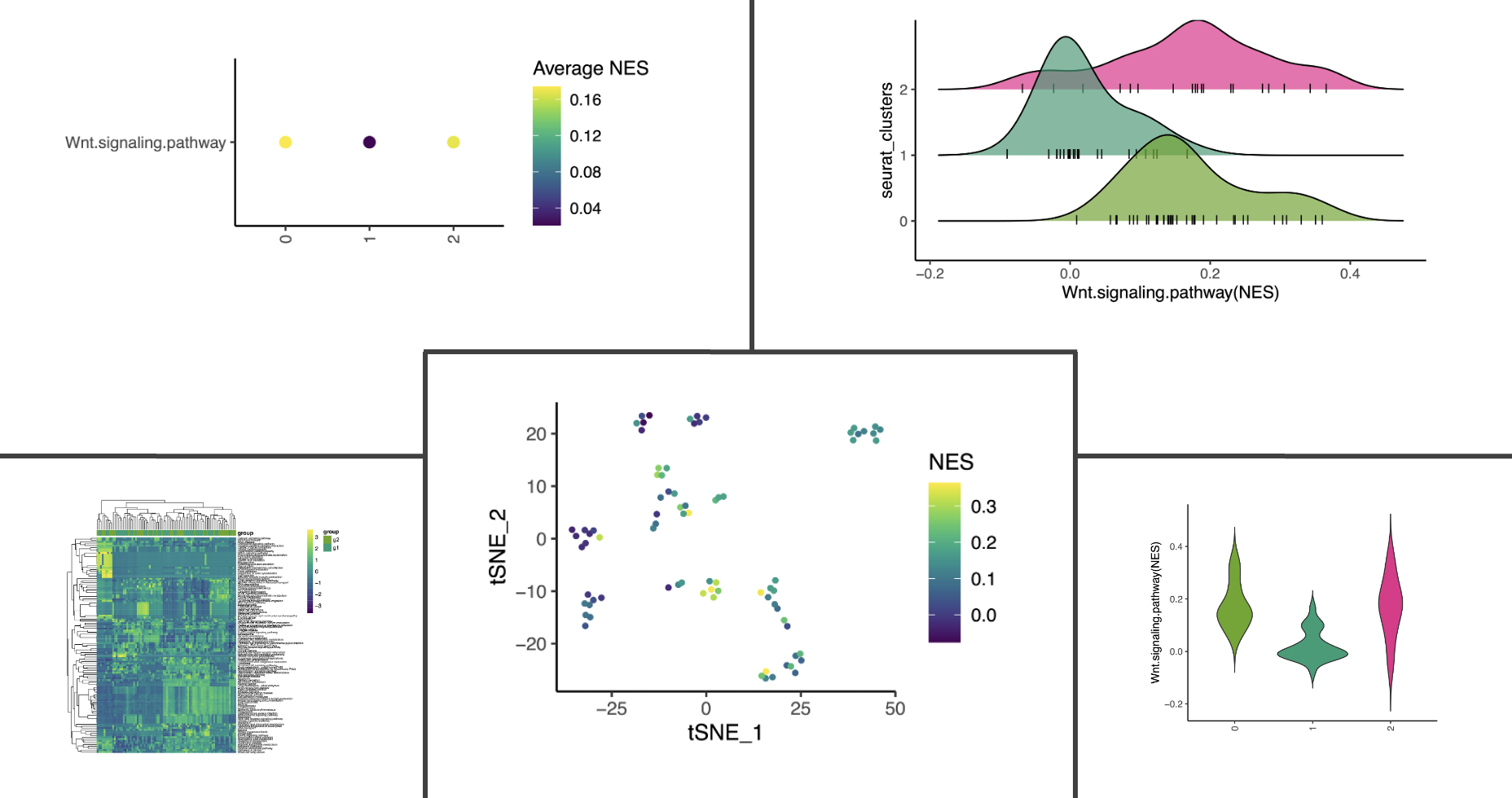
单细胞数据分析里面最基础的就是降维聚类分群,参考前面的例子:人人都能学会的单细胞聚类分群注释 ,这个大家基本上问题不大了,使用seurat标准流程即可,不过它默认出图并不好看,详见以前我们做的投票:可视化单细胞亚群的标记基因的5个方法,下面的5个基础函数相信大家都是已经烂熟于心了:
- VlnPlot(pbmc, features = c(“MS4A1”, “CD79A”))
- FeaturePlot(pbmc, features = c(“MS4A1”, “CD79A”))
- RidgePlot(pbmc, features = c(“MS4A1”, “CD79A”), ncol = 1)
- DotPlot(pbmc, features = unique(features)) + RotatedAxis()
- DoHeatmap(subset(pbmc, downsample = 100), features = features, size = 3)
其实pyscenic的转录因子分析结果也是一种打分,就可以去跟我们的降维聚类分群结合起来进行5种可视化展示。
感兴趣的可以申请 ,详见:https://experts.umich.edu/9854-kai-guo