我一直强调:数据挖掘的核心是缩小目标基因!
各种数据挖掘文章本质上都是要把目标基因集缩小,比如表达量矩阵通常是2万多个蛋白编码基因,不管是表达芯片还是RNA-seq测序的,采用何种程度的差异分析,最后都还有成百上千个目标基因。如果是临床队列,通常是会跟生存分析进行交集,或者多个数据集差异结果的交集,比如:多个数据集整合神器-RobustRankAggreg包 ,这样的基因集就是100个以内的数量了,但是仍然有缩小的空间,比如lasso等统计学算法,最后搞成10个左右的基因组成signature即可顺利发表。其实还有另外一个策略方向,有点类似于人工选择啦,通常是可以往热点靠,比如肿瘤免疫,相当于你不需要全部的两万多个基因的表达量矩阵进行后续分析,仅仅是拿着几千个免疫相关基因的表达矩阵即可。最近比较热门的有:自噬基因,铁死亡,EMT基因,核受体基因家族,代谢基因。还有一个最搞笑的是m6a基因的策略,完全是无厘头的基因集搞小,纯粹是为了搞小而搞小。
目前单细胞转录组大行其道,所以很多人喜欢使用公共的单细胞转录组数据集来缩小基因范围。学员在微信交流群分享了一个2024年5月的单细胞数据挖掘文章,标题是:《Single-cell combined with transcriptome sequencing to explore the molecular mechanism of cell communication in idiopathic pulmonary fibrosis》,研究者们重新分析了 GSE122960 这个单细胞转录组数据集,主要是第一层次降维聚类分群后,提取了巨噬细胞的特异性基因,然后走了随机森林生存分析算法,得到了 five most related key genes (CD163, IFITM2, IGSF6, S100A14 and SOD3). 有了目标的5个基因就可以很方便的各种简单分析来强调他们的生物学意义。比如去跟PDCD1基因看相关性:
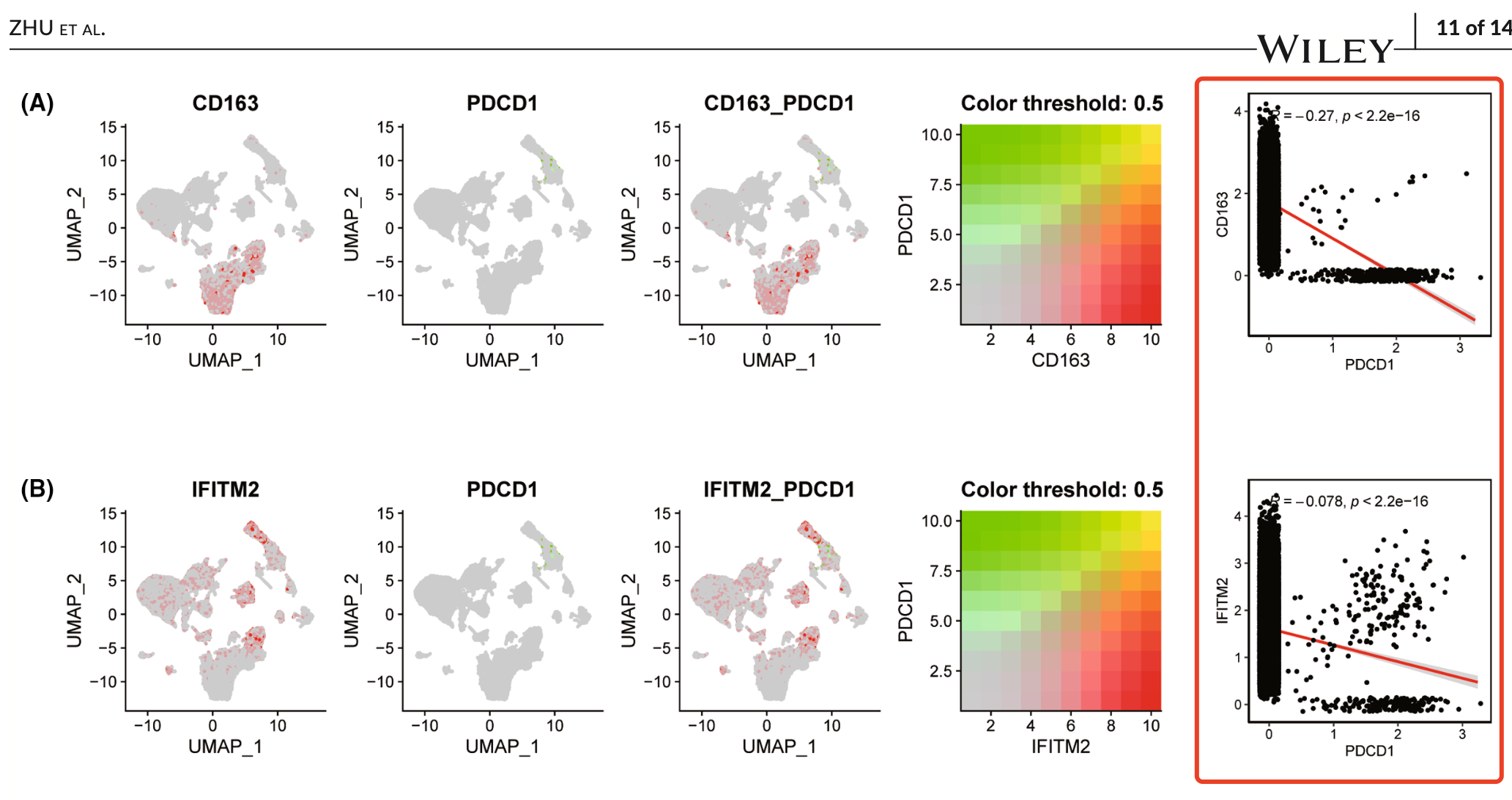
很明显,因为单细胞转录组数据有drop-out的特性,不能简单的拿原始的表达量矩阵去技术相关性啊,从散点图也可以看出来,每次需要计算相关性的两个基因都是各自在不同细胞里面大量表达量都是0,而这个0说不清楚是不是因为它真正的没有表达量还是说技术型的drop-out现象。
我问了ai有什么解决方案,它给出来了5个方案:
单细胞转录组数据中的dropout现象确实会对基因表达量的分析造成一定的影响。Dropout是指在单细胞测序过程中,某些基因在某些细胞中的表达量实际上不为零,但由于测序深度、扩增偏差等原因,在最终的表达矩阵中被记录为零。这会导致基因表达的相关性分析结果不准确。以下是几种处理这种情况的策略:
- 数据标准化:
- 在计算相关性之前,对数据进行标准化处理,以减少不同细胞之间的测序深度差异带来的影响。
- 使用非零表达量数据:
- 在计算相关性时,只使用表达量非零的数据点。这可以通过过滤掉表达矩阵中的零值来实现,但要注意这可能会减少可用数据量。
- 数据插补(Imputation):
- 对于dropout导致的零值,可以使用不同的插补方法来估计基因的真实表达水平。常见的插补方法包括基于均值、中位数的插补,或者更复杂的基于模型的插补方法,如MAGIC、scImpute等。
- 使用鲁棒的统计方法:
- 选择对零膨胀数据更鲁棒的相关性度量方法,如Spearman相关系数,它对异常值和非线性关系不那么敏感。
- 创建Metacells:
- Metacells是通过聚类等方法将相似的单细胞合并成更大的单元,这可以减少dropout的影响,因为dropout事件在合并后的单元中可能会被平均掉。
在实际操作中,可能需要根据数据的特点和研究目的,结合以上几种方法来处理dropout问题。例如,可以先进行数据插补,然后使用非零数据进行相关性分析,同时采用鲁棒的统计方法来提高分析的准确性。重要的是要对所使用的方法进行验证,并在可能的情况下,通过实验或其他数据源来支持分析结果。
如果大家的单细胞数量足够多,其实比较推荐Metacells方法,备选项就是Imputation啦,但是Imputation算法实在是太多了。而且面对10x这样的单细胞转录组技术有95%的0值,大多数Imputation算法表现差强人意。