提到蛋白质数据库,大家都很熟悉了,基本上每个数据库都会配备一个网页工具让大家查询或者做一些简单的蛋白质数据分析,以下是一些知名的蛋白质组数据库:
- TrEMBL:
由欧洲生物信息学研究所(EMBL-EBI)维护的蛋白质数据库,提供基于UniProt程序的自动注释信息。 - UniProt:
一个全面的、高质量的蛋白质数据库,包含来自UniProtKB/Swiss-Prot的手动注释蛋白质和来自TrEMBL的计算机注释蛋白质。 - Human Protein Atlas:
一个项目,旨在分析所有人类蛋白质在各种组织中的表达和定位。 - STRING:
一个数据库和网络资源,提供已知和预测的蛋白质-蛋白质相互作用。
但是蛋白质组数据库并不是蛋白质数据库,主要是ProteomeXchange联盟,它是一个开放的、公共的数据存储平台,专门用于存储和共享质谱(MS)数据。它由多个蛋白质组学数据存储库组成,包括PRIDE Archive、MassIVE、PeptideAtlas 以及iProX等 。
比如我们可以看到一个蛋白质组学文章:https://www.sciencedirect.com/science/article/pii/S0300483X20302912?via%3Dihub
Availability of data and material
The proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD020248.
ProteomeXchange的目的是促进数据的标准化、共享和再利用,支持蛋白质组学研究的进一步发展。它遵循FAIR原则(可查找性、可访问性、互操作性和可重用性),以确保数据的质量和可用性。一般来说我们熟悉iProX(国际蛋白质组学交流平台)和PRIDE(蛋白质组学鉴定数据库)即可:
- iProX: iProX(国际蛋白质组学交流平台)是一个由中国蛋白质组学研究者建立的数据库,旨在存储和分享基于质谱的蛋白质组学数据。iProX为科研人员提供了一个平台,用于提交、管理和访问蛋白质组学数据,支持数据的标准化和共享。iProX数据库也遵循ProteomeXchange的数据标准,促进了全球蛋白质组学数据的整合和分析。
- PRIDE: PRIDE(蛋白质组学鉴定数据库)是EMBL-EBI(欧洲生物信息学研究所)维护的一个数据库,它是ProteomeXchange联盟的一部分。PRIDE专门收集和存储质谱数据,特别是蛋白质和肽段的鉴定和定量信息。PRIDE数据库支持用户上传数据、下载公开数据集,并提供了一系列的分析工具,帮助科研人员进行蛋白质组学研究。
PRIDE(蛋白质组学鉴定数据库)
是EMBL-EBI(欧洲生物信息学研究所)维护的一个数据库,可以通过链接:https://www.ebi.ac.uk/pride/archive?sortDirection=DESC&page=2&pageSize=20
查看数据库目前有的数据集数量: List of Datasets (27273)
比如最近的一个公共数据集就是:Neutrophil-derived migrasomes are an essential part of the coagulation system, mouse.
可以很清晰的看到这个pride数据库为这个数据集提供了raw格式的质谱仪器数据,以及蛋白质组表达量矩阵文件:

详细的数据集链接在:https://www.ebi.ac.uk/pride/archive/projects/PXD051229
不过我们一般来说就打开里面的ms220042-dia-20220111.xls文件进行后续的蛋白质组表达量差异分析即可
iProX(国际蛋白质组学交流平台)
在 https://www.iprox.cn/page/BWV016.html 可以看到就 3,676 entries,因为是一个由中国蛋白质组学研究者建立的数据库,所以绝大部分数据集都是中国科研工作者提供的,而且绝大部分都是在PRIDE(蛋白质组学鉴定数据库)也有一个id,如下所示;
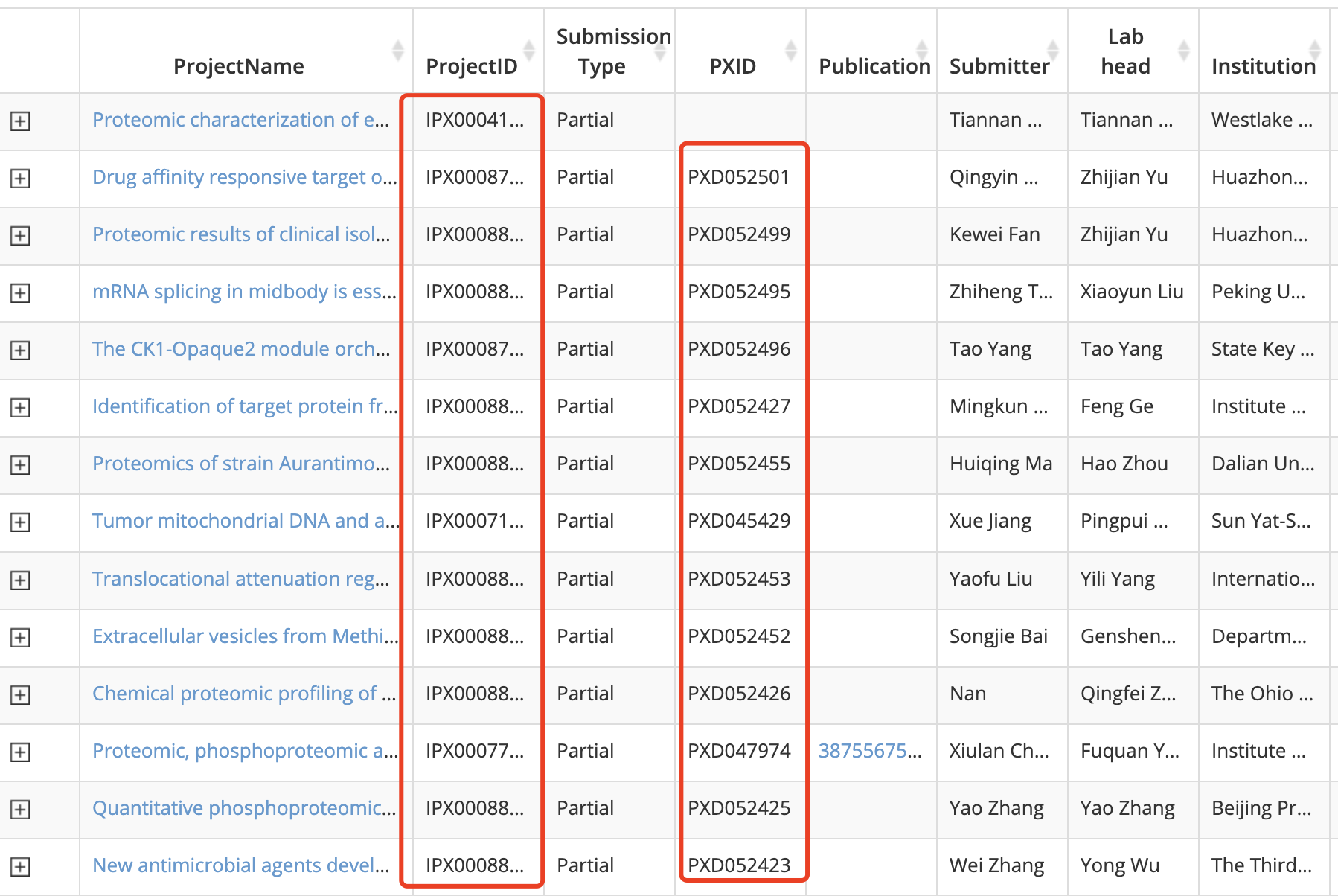
任意点击一个数据集进去:https://www.iprox.cn/page/ProjectFileList.html?projectId=IPX0006535000
Proteome of Chinese Breast Cancers (FUSCC-Shao Lab)
IPX0006535000
Partial
PXD042886
Jiang YZ, Shao ZM. Molecular features and clinical implications of the heterogeneity in Chinese patients with HER2-low breast cancer. Nature Communications. 2023 Aug 22;14(1):5112-. doi:10.1038/s41467-023-40715-x.
Zhiming Shao
Zhiming Shao
Fudan University Shanghai Cancer Center
1
2023-06-11 08:40:12
也可以看到它的其它id,只需要有id就可以访问它在不同数据库里面的数据记录,比如:
- https://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD042886
- https://www.ebi.ac.uk/pride/archive/projects/PXD042886
如果数据集并没有提供蛋白质表达量矩阵文件
因为本来就是有很多不同的蛋白质组学技术平台,比如 DIA、Label Free和TMT标记,它们的各自的 数据预处理流程 也有点区别哦:
DIA(Data-Independent Acquisition)数据预处理流程:
-
Spectronaut搜库策略:
- 使用Spectronaut软件进行搜库,获取肽段和蛋白的相对定量信息。
- 数据log2转换、缺失值过滤和填充、数据标准化,可能使用Combat去除批次效应。
-
DIA-NN搜库策略:
- 使用DIA-NN进行搜库,得到相对定量值。
- 进行log2转换、数据标准化、缺失值处理,最后鉴定差异蛋白。
Label Free数据预处理流程:
-
Maxquant搜库策略:
- 搜库结果提供Intensity、iBAQ、LFQ intensity三种定量值。
- 数据log2转换、样本内中值或quantile标准化、缺失值过滤和填充。
- 进行差异定量分析。
-
Proteome Discoverer(PD)搜库策略:
- 默认定量值为iBAQ。
- 标准化方式为FOT(Fraction of Total)。
- 缺失值填充,选择合适阈值进行填充。
- 下游数据分析。
TMT(Tandem Mass Tag)数据预处理流程:
- MSFragger搜库策略:
- 使用MSFragger进行搜库,得到pepXML格式搜库结果文件。
- 利用Philosopher工具包进行肽段、蛋白和翻译后修饰的定量和过滤。
- PeptideProphet进行肽段鉴定和验证,PTMProphet进行修饰位点鉴定。
- ProteinProphet用于蛋白鉴定。
- 使用Philosopher进行FDR过滤和定量,获得TMT reporter ion intensity。
- 参考通道样品校正,进行多重数据转换和标准化。
- log2转换、样本内中值标准化、缺失值处理、批次效应去除、差异表达分析。