看到朋友圈好多人转发了一个最新的胃癌单细胞研究,数据集是GSE239676,在GEO界面可以看到如下所示的文件:
5.4M Aug 15 23:21 GSE239676_barcodes.tsv.gz
939M Aug 15 23:26 GSE239676_count_matrix.mtx.gz
53K Aug 15 23:32 GSE239676_features.tsv.gz
5.5M Aug 15 23:21 GSE239676_meta.tsv.gz
2024年8月1日,美国德克萨斯大学王凌华、Jaffer A. Ajani、Pawel K. Mazur、中国科学院基础医学与肿瘤研究所覃江江共同通讯在Gastroenterology(IF25.7)在线发表了题为“Atlas of metastatic gastric cancer links ferroptosis to disease progression and immunotherapy response”的论文,研究揭示了转移性胃癌的细胞和分子机制,并提出了一种靶向铁死亡防御机制联合CAR-T细胞疗法的治疗策略。
可以看到,作者给出来的降维聚类分群的结果还不错:

但是我自己在跑上面的代码的时候就发现了一个小瑕疵,如下所示,根据上面的基因很容易给出来各个单细胞亚群的名字,包括CD4和CD8的T细胞,髓系免疫细胞,上皮细胞,b细胞和浆细胞,甚至还有树突细胞:
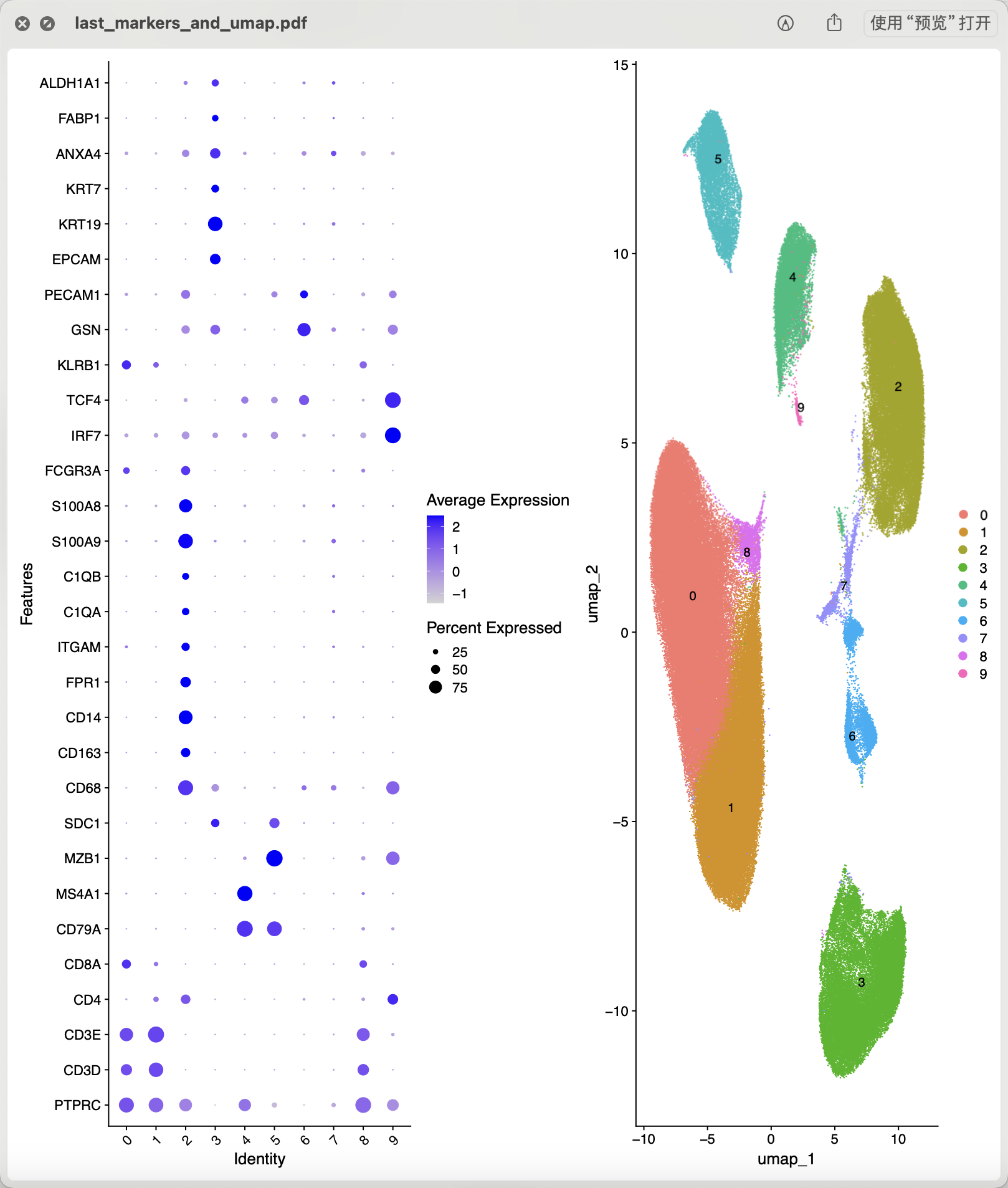
但是stromal就好奇怪,刚开始以为是分辨率的问题。本来呢,基质细胞主要包括以下几种类型:
- 成纤维细胞(Fibroblasts):在肿瘤微环境中,成纤维细胞可以分化为癌症相关成纤维细胞(Cancer-Associated Fibroblasts, CAFs),它们通过分泌多种细胞因子和细胞外基质(ECM)成分,促进肿瘤的发展和转移。
- 内皮细胞(Endothelial Cells):内皮细胞在肿瘤血管生成中起到关键作用,它们可以形成新的血管,为肿瘤提供氧气和营养。
- 平滑肌细胞(Smooth Muscle Cells, SMCs):在某些情况下,平滑肌细胞也存在于肿瘤微环境中,尤其是在血管结构中。
- 周细胞(Pericytes):周细胞是血管壁的一部分,它们与内皮细胞相互作用,参与血管的稳定和成熟。
其中内皮细胞主要是区分成为了淋巴内皮和血管内皮, 其中血管可以细分为动脉静脉和毛细血管:
- lymphatic ECs (LECs; CCL21, PROX1).
- arteries (HEY1, IGFBP3), capillaries (CD36, CA4), veins (ACKR1)
但是LUM和DCN等基因居然是缺失的,导致我如下所示的代码就失效了;
library(ggplot2)
genes_to_check =list(
CM=c("TTN","MYH7","MYH6","TNNT2") ,
EC=c("VWF", "IFI27", "PECAM1","MGP"),
FB=c("DCN", "C7" ,"LUM","FBLN1","COL1A2"),
MP=c("CD163", "CCL4", "CXCL8","PTPRC"),
SMC=c("ACTA2", "CALD1", "MYH11"),
Tc=c("CD3D","CD3E"),
peric=c("ABCC9","PDGFRB","RGS5")
)
genes_to_check = lapply(genes_to_check,function(x){
unique( c(str_to_title(x),str_to_upper(x)))
} )
p_all_markers=DotPlot(sce.all.int,
features = genes_to_check,
group.by = 'RNA_snn_res.0.1',
scale = T,assay='RNA' ) +
theme(axis.text.x=element_text(angle=45,hjust = 1))
p_all_markers
这个时候我就明白了其实是表达量矩阵文件的问题,居然是 8630个基因,然后是 222240个细胞 。正常情况下, 有两三万个基因才对啊!大家可以自己下载这些文件走一下我们的标准的降维聚类分群,就明白了;
if(T){
library(data.table)
library(Matrix)
# https://hbctraining.github.io/scRNA-seq/lessons/readMM_loadData.html
#
mtx=readMM( "inputs/GSE239676_count_matrix.mtx.gz" )
mtx[1:4,1:4]
dim(mtx)
cl=fread( "inputs/GSE239676_barcodes.tsv.gz" ,
header = F,data.table = F )
head(cl)
rl=fread( "inputs/GSE239676_features.tsv.gz" ,
header = F,data.table = F )
head(rl)
dim(rl)
dim(mtx)
colnames(mtx)=cl$V1
rownames(mtx)=rl$V1
mtx[1:4,1:4]
dim(mtx)
meta=fread( "inputs/GSE239676_meta.tsv.gz" ,
header = T,data.table = F )
head(meta)
rownames(meta)=colnames(mtx)
library(Seurat)
sce.all=CreateSeuratObject(counts = mtx ,
meta.data = meta,
min.cells = 5,
min.features = 300 )
as.data.frame(sce.all@assays$RNA$counts[1:10, 1:2])
head(sce.all@meta.data, 10)
table(sce.all$orig.ident)
}
这些年陆陆续续处理了一两千个单细胞转录组公共数据集了,总是会碰到一些缺胳膊断腿的。有一些数据集,比如GSE233447就完全不给任何基因名字,详见:细胞名字可以不给但是基因名字不能不要啊。而这个美国德克萨斯大学王凌华老师的新鲜出炉的胃癌单细胞给一半的基因的表达量矩阵,这个操作也是有一点点迷惑。。。
看起来也是需要去下载fastq文件然后自己走cellranger流程啊:https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1000548