交流群有小伙伴分享了一个新鲜出炉的2024的单细胞转录组配合空间单细胞的文献:《Single-cell and spatial transcriptomics analysis of non-small cell lung cancer》,可以看到这个研究的细胞数量还是很可观的,接近100万啦 :
- 503,549 were from tumour
- 392,257 from combined background and healthy tissue
但是实际上的病人数量并不多: lung tissue resections from 25 treatment-naive patients with either LUAD (n = 13), LUSC (n = 8) or undetermined lung cancer (LC, n = 4), and two healthy deceased donors
也就是说这25个病人每个人都是癌症和癌旁两个样品 :We collected both tumour and matched normal non-tumorigenic tissue (i.e., background), isolated CD45+ immune cells ,总计50个左右的样品居然有90多万个细胞数量,相当于是每个样品都是两万多的细胞数据啦。但是每个样品搞这么多细胞干什么啊?
探究一下这个单细胞转录组数据集是E-MTAB-13526
可以很容易下载到每个样品的表达量矩阵文件,然后很简单的每个样品独立的读取,可以看到基本上每个10x技术的单细胞转录组样品的细胞数量都是在1万附近,但是有极个别是4万左右的细胞数量,这个就很恐怖了!
> do.call(rbind,lapply(sceList, dim))
[,1] [,2]
P1_B1 15102 2324
P10_B1 14979 1925
P11_B1 16806 3048
P12_B1 11596 307
P13_B1 16601 4686
P14_B1 10142 257
P15_B1 17798 6924
P15_B2 19390 8722
P16_B1 17928 13081
P16_B2 19241 21566
P17_B1 15597 3674
P17_B2 20841 15612
P18_B1 21455 39755
P18_B2 22714 34077
P19_B1 16663 4954
P2_B1 15175 1694
P20_B1 21419 37558
P20_B2 23602 40075
P21_B1 19808 25359
P21_B2 16102 2014
P21_B3 15346 3943
P22_B1 22948 62387
P22_B2 15137 2873
P23_B1 21809 17545
P23_B2 17210 6710
P24_B1 23575 40268
P24_B2 17609 9289
P3_B1 18063 4223
P4_B1 17080 2863
P4_B2 21807 18010
P4_B3 21944 22421
P5_B1 16831 3466
P6_B1 14076 1085
P7_B1 13038 665
P8_B1 18004 7939
P8_B2 22034 47517
P9_B1 15712 10039
如果是针对上面的数据进行降维聚类分群,会发现图很丑很丑,文献里面的二维UMAP就丑爆了:
只需要简单的抽样即可解决这个问题
上面的列表里面的是多个独立的单细胞Seurat对象,所以需要首先merge然后JoinLayers,
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ] )
# 26047 features across 528855 samples within 1 assay
sce.all <- JoinLayers(sce.all)
sce.all = subset(sce.all,downsample=10000)
# 26047 features across 233585 samples within 1 assay
可以看到之前是528855细胞数量,简单的抽样后就是233585个细胞啦,然后出图也看起来OK:
文章的核心发现可能是错误的?
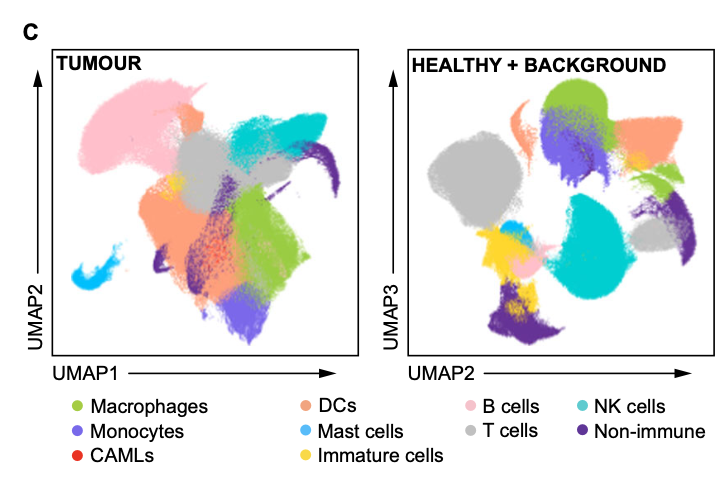
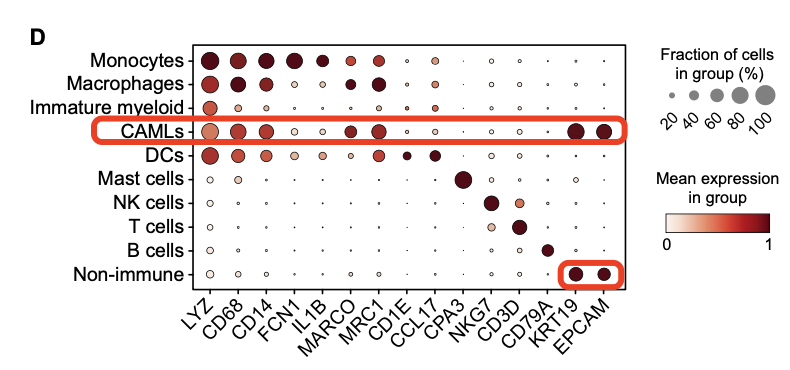
该文章最后的落脚点是 cancer-associated macrophage-like cells (CAMLs),它的特殊性在于 co-expression of myeloid (LYZ, CD68, CD14, MRC1) and epithelial genes (KRT19, EPCAM) ,如下所示:
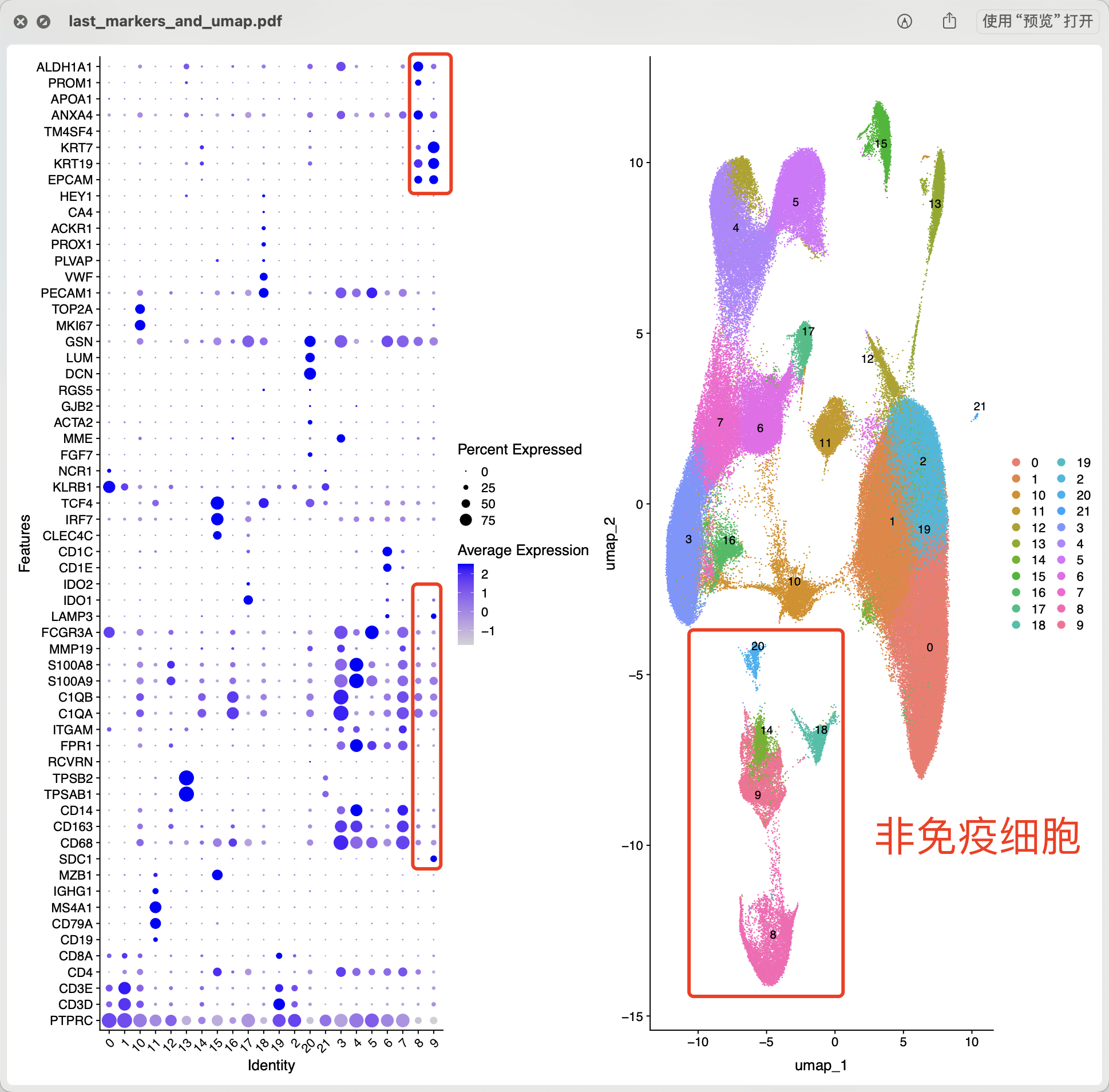
但是它的单细胞转录组数据质量如此差,我们虽然说是只需要简单的抽样后让它的降维聚类分群看起来没有那么丑,但是不能从源头解决它的质量问题。首先我们看看 normal non-tumorigenic tissue (i.e., background), isolated CD45+ immune cells,因为是流式细胞筛选技术,所以里面肯定是有一些非免疫细胞,比如18是内皮细胞,20是成纤维,然后8,9,14确实是上皮细胞但是里面有一些巨噬细胞的特异性基因的混入 :
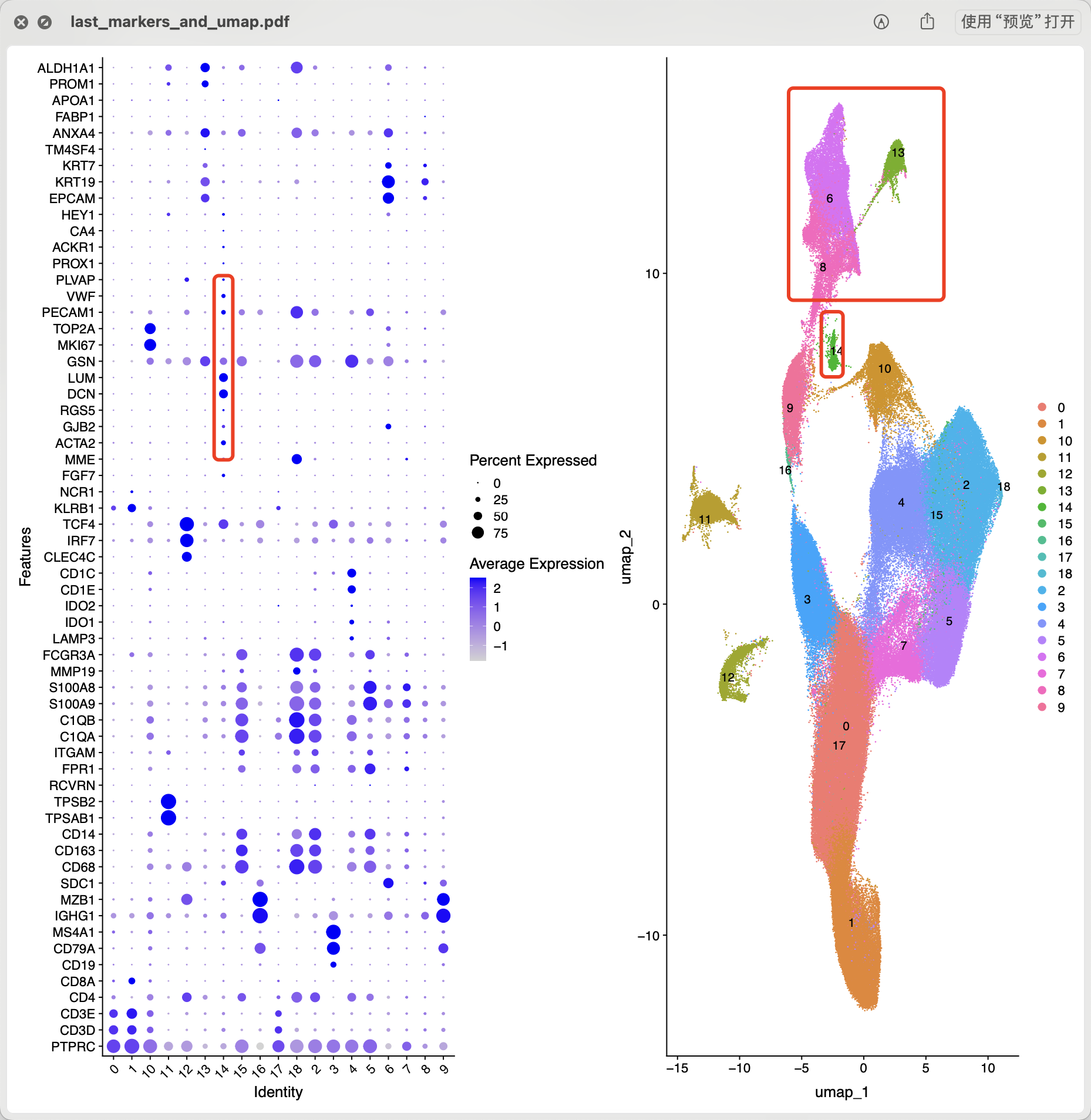
再看看肿瘤样品部分,同样的也是流式细胞技术筛选了免疫细胞,但是会有漏网之鱼,如下所示其中14是内皮细胞和成纤维的混合,然后 6,8,13都是有巨噬细胞特性的上皮细胞 :
让我们再回顾一下这个名字,cancer-associated macrophage-like cells (CAMLs),是不是有一点搞笑啊, 它为什么在癌症和癌旁里面都有呢?很明显的就是这个文章的作者们的实验特殊性导致的。