不同格式的单细胞表达量矩阵文件读取的分门别类的代码我们都分享了好多次,最后都是要成为 Seurat对象。详见:读取不同格式的单细胞转录组数据及遇到问题的解决办法,简单的汇总一下就是:
- h5格式的单细胞文件读取:
- 使用
Seurat包中的Read10X_h5函数。
- 使用
- 10X格式的单细胞文件读取:
- 10X格式通常包含
matrix.mtx.gz、genes.tsv.gz(或features.tsv.gz)和barcodes.tsv.gz三个文件。 - 使用
Seurat包中的Read10X函数。
- 10X格式通常包含
- txt.gz格式文件读取:
- 使用
data.table包中的fread函数。
- 使用
- csv格式文件读取:
- 同样使用
data.table包中的fread函数。
- 同样使用
遇到的问题及解决办法:
- 非标准10X数据集:有时GEO数据库上传的数据可能不符合标准格式,需要进行预处理。
- 去除第一行和第一列:如果第一行或第一列包含非数据内容,需要去除以避免读取错误。
- 解决办法:使用R的数据处理函数,如
read.table时指定quote = ""和row.names = 1,或者手动去除不需要的行列。
- 解决办法:使用R的数据处理函数,如
-
特征(feature)列问题:如果特征列不符合预期,可能需要指定
gene.column参数。 -
使用
read.table:有评论建议使用read.table函数,并通过设置参数quote = ""和row.names = 1来避免切割文件。
其它:比如.loom文件可以使用Seurat包中的Read10X_loom函数或者loomR包来读取。
一个让人脑子有坑的公共数据集
正常情况下,任意公共数据集,都是只需要把表达量矩阵文件读入到r编程语言里面的成为了一个合格的Seurat对象,后续的降维聚类分群等常规单细胞转录组数据分析基本上可以套用我们的标准代码即可,
但是最近交流群有小伙伴反馈了一个新鲜出炉的文章:《Single-cell profiling reveals altered immune landscape and impaired NK cell function in gastric cancer liver metastasis》它的数据集是GSE246662,在GEO界面可以看到作者给出来的是如下所示的文件 :
21M 11 1 2023 GSM7874169_HL1.csv.gz
22M 11 1 2023 GSM7874170_HL2.csv.gz
16M 11 1 2023 GSM7874171_HL3.csv.gz
13M 11 1 2023 GSM7874172_GC1.csv.gz
22M 11 1 2023 GSM7874173_GC2.csv.gz
33M 11 1 2023 GSM7874174_GC3.csv.gz
17M 11 1 2023 GSM7874175_LM1.csv.gz
27M 11 1 2023 GSM7874176_LM2.csv.gz
25M 11 1 2023 GSM7874177_LM3.csv.gz
如果是每个文件当做是同样的格式批量读取,就会出现如下所示的奇怪的降维聚类分群图:
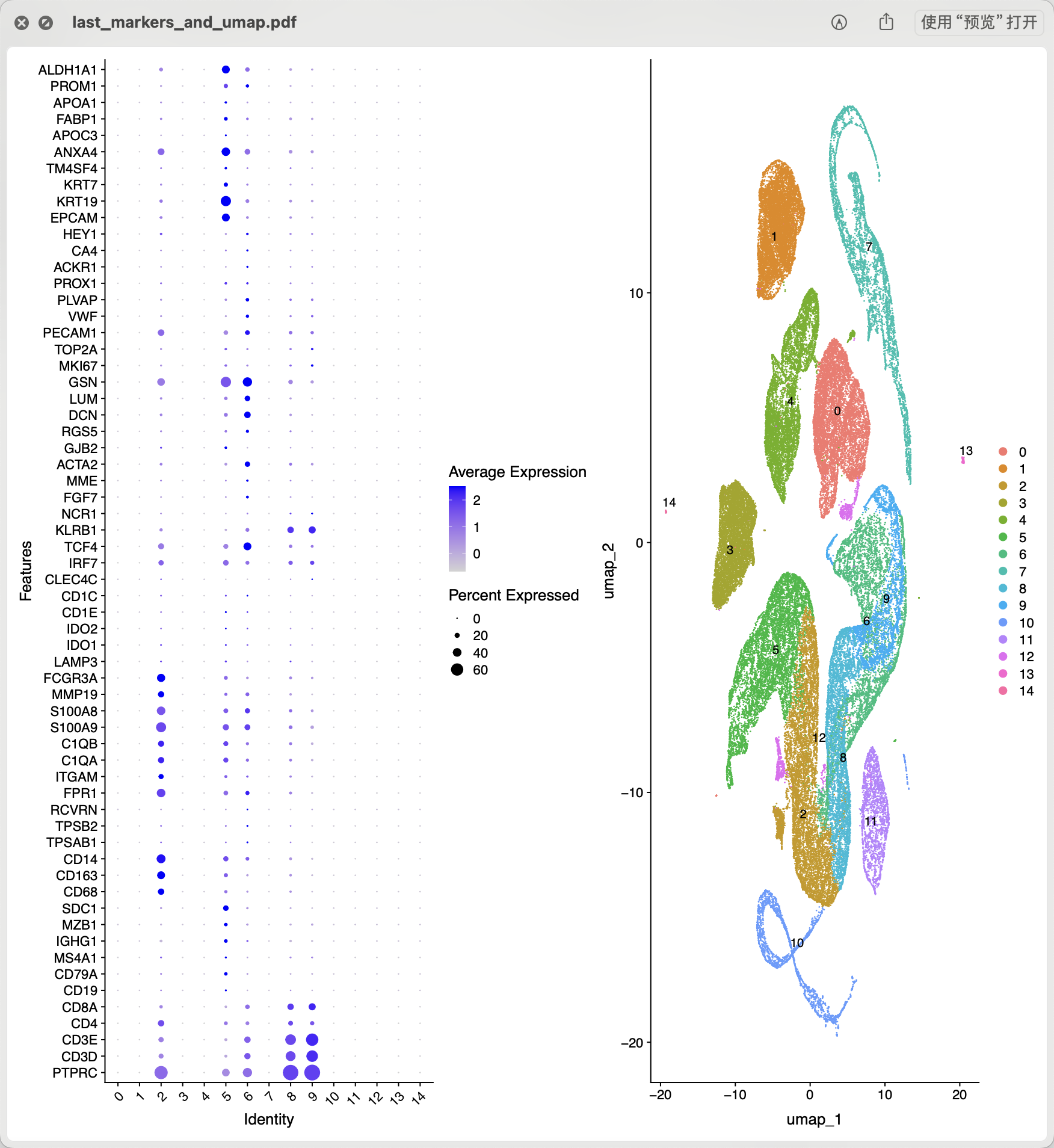
也就是说,0,1,3,4这4个亚群,它们没有任意的标记基因,但是确实是在UMAP的二维图上面很清晰的聚成为了一个独立的很清晰的cluster,我打打开了这些亚群的top3基因才发现问题所在:
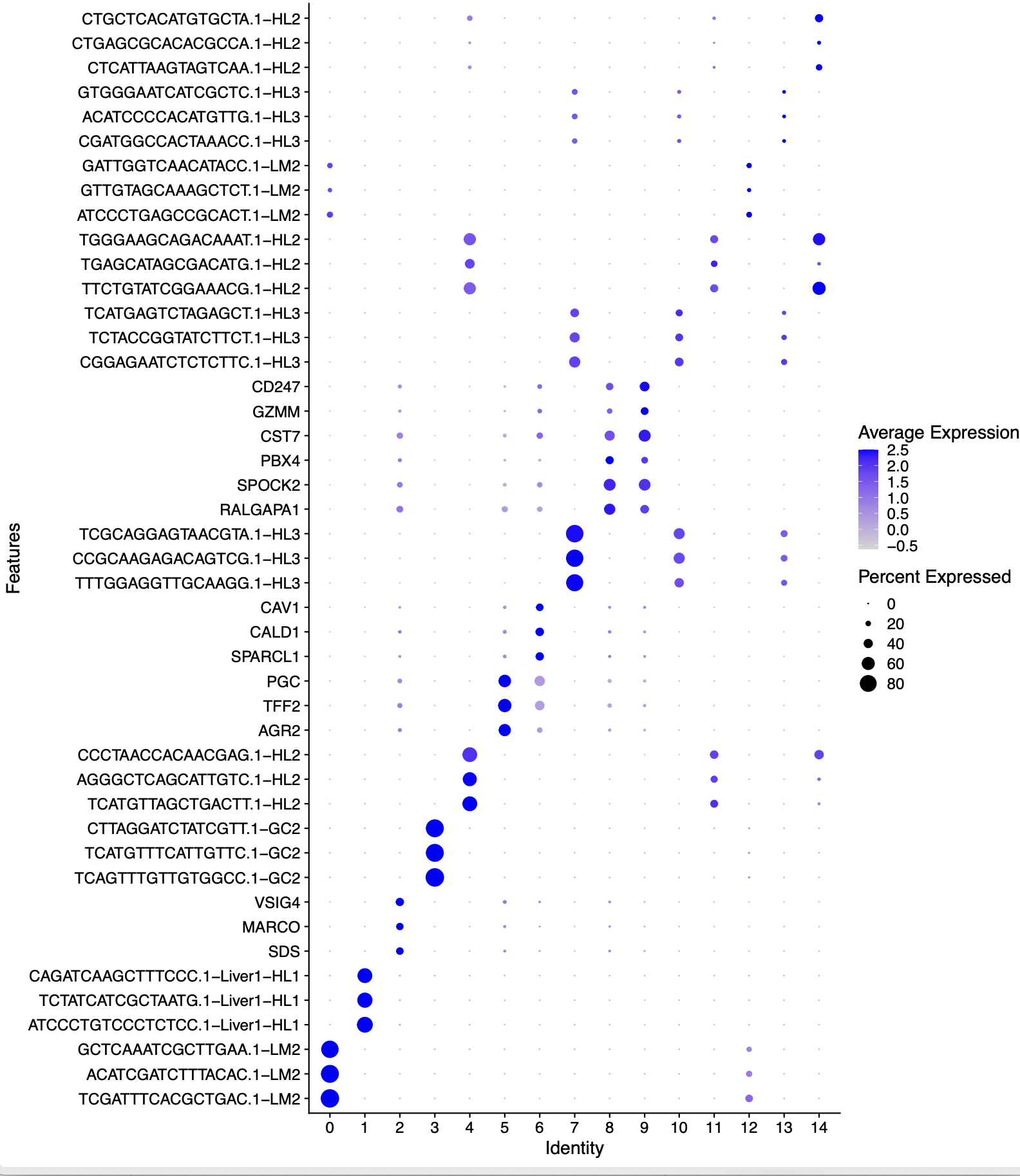
有一些样品居然是被转置的,正常的表达量矩阵应该是列是细胞,然后行是基因。
所以需要修改我们的默认读取表达量矩阵的代码,如下所示:
dir='GSE246662_RAW'
samples=list.files( dir )
samples
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
ct=data.table::fread( file.path(dir,pro ) ,data.table = F)
ct[1:4,1:4]
rownames(ct)=ct$V1
ct=ct[,-1]
if( ! sum(nchar(colnames(ct)) > 12) > 1000 ){ ct= t(ct)}
sce =CreateSeuratObject(counts = ct ,
project = gsub('.csv.gz','',pro) ,
min.cells = 5,
min.features = 300 )
return(sce)
})
do.call(rbind,lapply(sceList, dim))
因为基因名字大多数长度小于10的,但是细胞的名字都是大于10的,所以我加上了一个很简单的判断。就可以得到一个正常的Seurat对象然后降维聚类分群啦, 如下所示的结果,其中那个merge亚群其实是可以细分,本来就是肝细胞为主但是里面不知道为什么混入了t细胞的基因,所以我没有给文章那这样的肝细胞这样的名字。
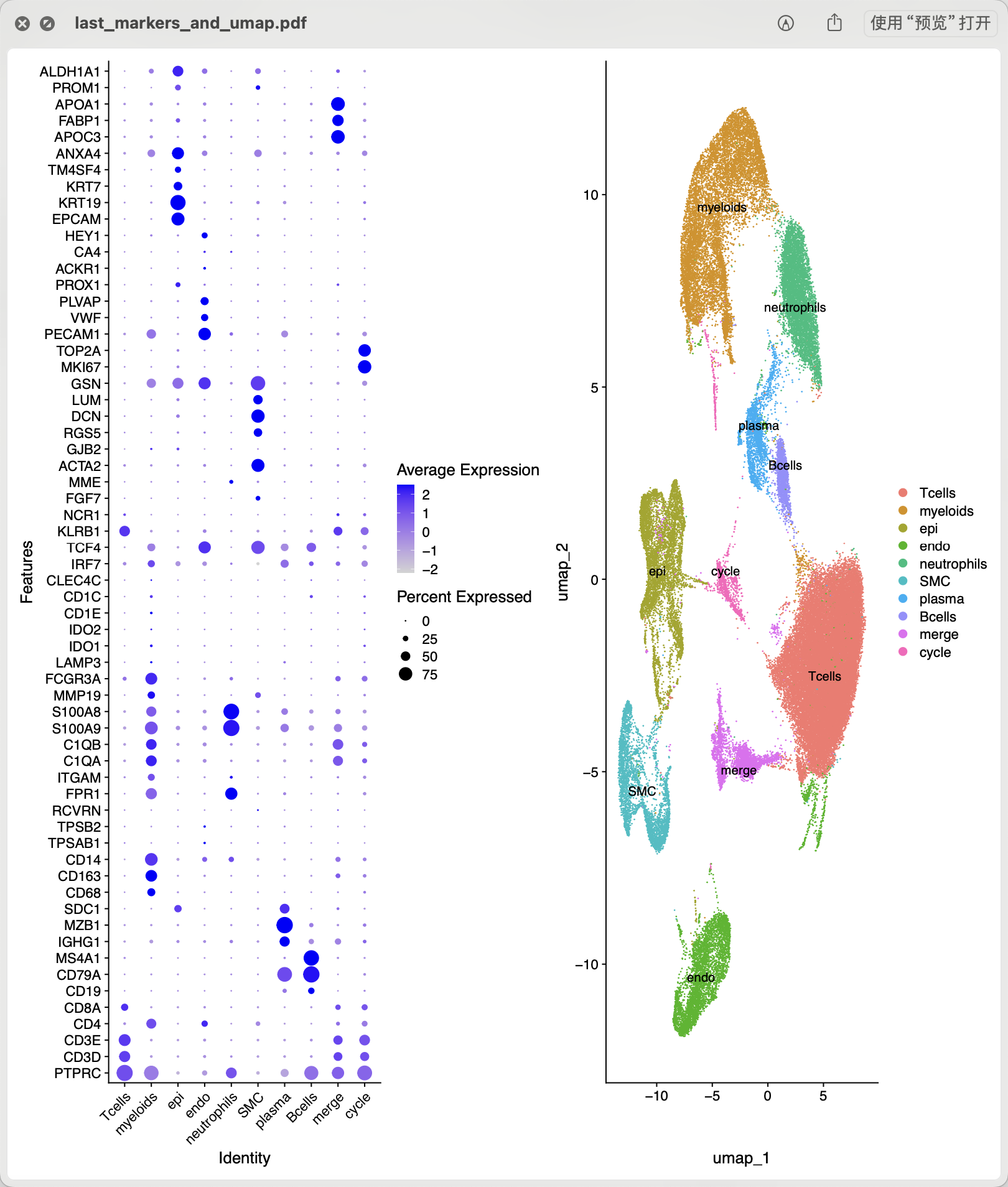
虽然说是跟文章有点差异,但是这个出入是可以接受的:

学徒作业
完成这个数据集,GSE246662,的降维聚类分群后,看看里面的肝细胞是不是真的有t淋巴细胞的特性。毕竟新鲜出炉的2024的单细胞转录组配合空间单细胞的文献:《Single-cell and spatial transcriptomics analysis of non-small cell lung cancer》,就是提出来了 cancer-associated macrophage-like cells (CAMLs),它的特殊性在于 co-expression of myeloid (LYZ, CD68, CD14, MRC1) and epithelial genes (KRT19, EPCAM) ,也算是另辟蹊径?