看到了一个2022的文章,标题是:《Bromodomain Inhibitors Modulate FcgR-Mediated Mononuclear Phagocyte Activation and Chemotaxis》。这个研究里面的转录组数据是通过RNA测序(RNA-seq)技术获得的,这些数据来自两个不同的细胞类型:树突细胞(Dendritic Cells, DCs)和巨噬细胞(Macrophages),以及它们在不同实验条件下的反应。
不同的细胞类型就是不同的数据集,每个数据集内部都是4个分组:
- IC(Immune Complexes)刺激:树突细胞被免疫复合物刺激,这是模拟体内免疫反应的一种实验条件。
- Ova(Ovalbumin)刺激:树突细胞被卵清蛋白刺激,这是一种常用的模型抗原,用于研究免疫细胞的激活。
- DMSO对照组:细胞未接受任何处理,DMSO是iBET药物的溶剂,这里作为阴性对照。
- iBET处理组:细胞被BET蛋白抑制剂iBET处理,用于研究iBET对树突细胞转录组的影响。
在(Gene Expression Omnibus, GEO)中,可以通过提供的访问号码GSE200033和GSE200226获取这两个数据集的表达量矩阵。
我们很容易读取树突细胞(BMDCs)的表达量矩阵,代码如下所示:
rm(list = ls())#清空当前的工作环境
options(stringsAsFactors = F)#不以因子变量读取
options(scipen = 20)#不以科学计数法显示
library(data.table)
data<-data.table::fread("GSE200226_CountTable_BMDC.csv.gz",
header = T,
data.table = F)
data=data[!duplicated(data$V1),]
mat<-floor(data[,c( 2:ncol(data))])
rownames(mat)= data[,1]
keep_feature <- rowSums (mat > 1) > 1
symbol_matrix <- mat[keep_feature, ]
rownames(symbol_matrix)=rownames(mat)[keep_feature]
dat=log(edgeR::cpm(symbol_matrix)+1)
然后简单的根据这个表达量矩阵的基因的SD值排序后挑选里面的排名靠前的1000个基因后绘制如下所示的热图:

很明显的可以看到这个表达量矩阵里面BET蛋白抑制剂iBET处理造成的表达量变化是最大的,然后是卵清蛋白刺激和IC的稍微小一点的差异,然后每个组里面的5个重复其实也略微有一点点分组,可能是先做了3个重复然后补了2个样品。
上面的热图代码是:
dat[1:4,1:4]
cg=names(tail(sort(apply(dat,1,sd)),1000))#apply按行('1'是按行取,'2'是按列取)取每一行的方差,从小到大排序,取最大的1000个
library(pheatmap)
pheatmap(dat[cg,],show_colnames =F,show_rownames = F) #对那些提取出来的1000个基因所在的每一行取出,组合起来为一个新的表达矩阵
n=t(scale(t(dat[cg,]))) # 'scale'可以对log-ratio数值进行归一化
n[n>2]=2
n[n< -2]= -2
n[1:4,1:4]
pheatmap(n,show_colnames =F,show_rownames = F)
ac=data.frame(group=group_list)
rownames(ac)=colnames(n)
pheatmap(n,show_colnames =F,show_rownames = F,
annotation_col=ac)
我们该如何判断这个矩阵确实是来自于树突细胞呢
前面我们提到了不同的细胞类型:树突细胞(Dendritic Cells, DCs)和巨噬细胞(Macrophages),是不同的数据集,有不同的表达量矩阵文件。但是我们如何相信作者给出来的文件是否准确无误呢?
其实很简单,需要靠生物学背景,比如任意单细胞转录组数据集里面都可以看到不同细胞亚群的特异性基因,如下所示:
SCP1661_meyloids_markers_list =list(
macrophages=c('Adgre1', 'Cd14', 'Fcgr3'),
cDCs=c('Xcr1', 'Flt3', 'Ccr7'),
pDCs=c('Siglech', 'Clec10a', 'Clec12a') ,
monocytes=c('Ly6c2' , 'Spn'),
neutrophils=c('Csf3r', 'S100a8', 'Cxcl3')
)
df = data.frame(
celltype=rep(names(SCP1661_meyloids_markers_list),
times=unlist(lapply(SCP1661_meyloids_markers_list, length))),
gene=unlist(SCP1661_meyloids_markers_list)
)
library(stringr)
df=df[df$gene %in% rownames(dat),]
cg=df$gene
这个时候,首先查看树突细胞数据集里面的各个髓系免疫细胞单细胞亚群的特异性基因的热图,这个时候我们不会选择横向基因层面归一化,而是直接可视化log的CPM值,如下所示 :
library(pheatmap)
#tmp=dat[cg,]
n=t(scale(t(dat[cg,]))) # 'scale'可以对log-ratio数值进行归一化
n[n>2]=2
n[n< -2]= -2
n[1:4,1:4]
pheatmap(n,show_colnames =F,show_rownames = F)
ac=data.frame(group=group_list)
ar=data.frame(gene=df$celltype)
rownames(ac)=colnames(n)
rownames(ar)=rownames(n)
pheatmap(tmp,show_colnames =F,show_rownames = T,
display_numbers = T,
annotation_row =ar,
annotation_col=ac)
如下所示的效果:
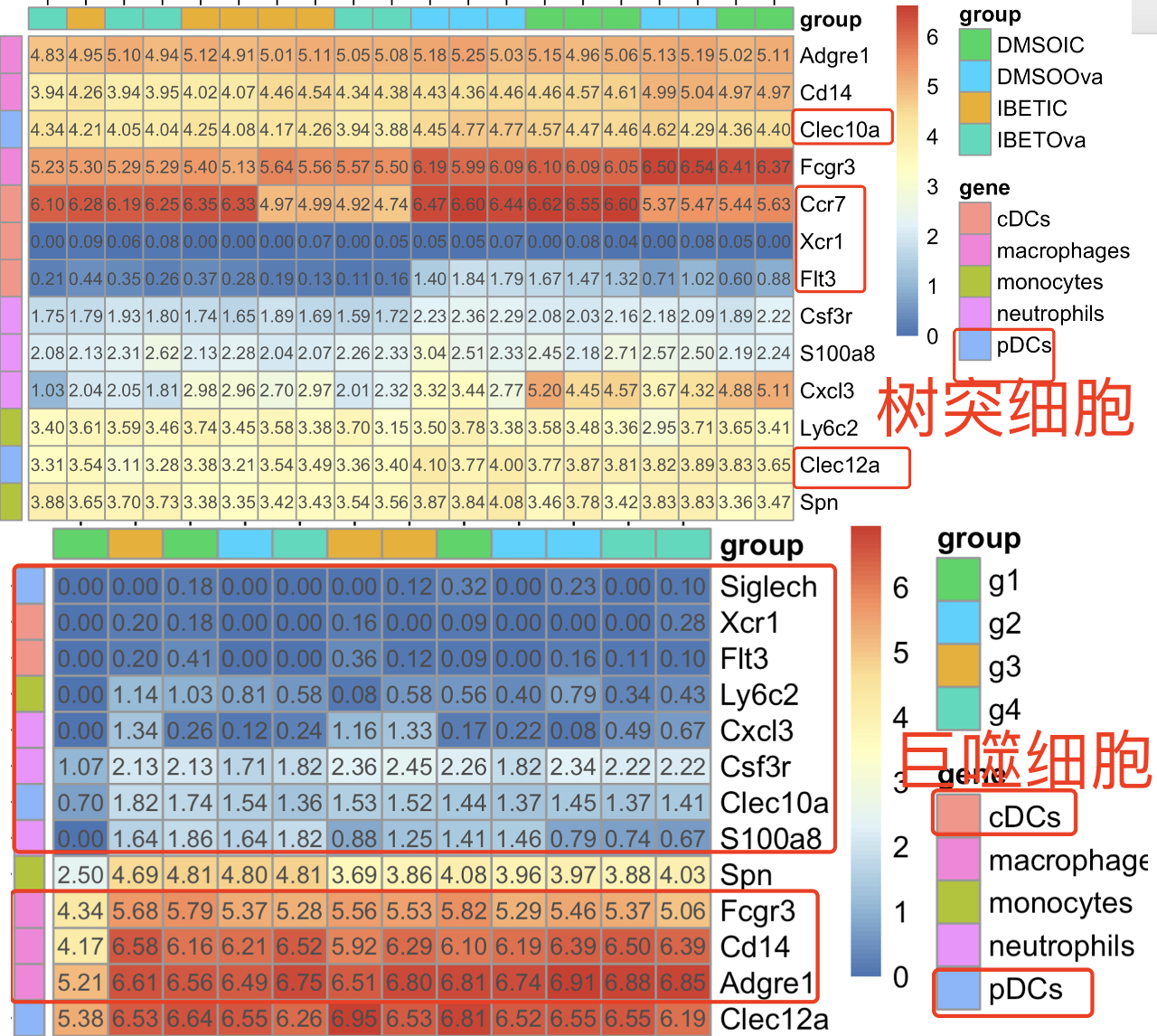
那么,为什么这个时候我们不会选择横向基因层面归一化呢,因为我想看的巨噬细胞和树突细胞特异性基因是否在每个数据集内部高表达,而不是在数据集内部对比那4个分组的表达量差异。
可以看到,在树突细胞数据集里面的Clec10a基因确实是表达量很高,这个巨噬细胞数据集里面是略低的。
学徒作业
正常情况下,应该是把上面的这两个矩阵合并后可视化,这个时候就可以继续横向基因层面归一化,可以看到具体的每个基因在两个数据集的差异来区分巨噬细胞和树突细胞数据集。
那,什么时候需要依据纵向的样品进行归一化呢?反正我基本上没有遇到过这样的需求,上面的热图就是没有选择横向基因层面归一化,而是直接可视化log的CPM值,其实也可以做依据纵向的样品的归一化,但是因为巨噬细胞和树突细胞特异性基因并不多,而且表达量差异悬殊,是否归一化就不那么重要了。