最近在咱们的微信交流群看到了小伙伴反馈一个数据挖掘文章从根上就错了,标题:《Integrative analyses of bulk and single-cell transcriptomics reveals the infiltration and crosstalk of cancer-associated fibroblasts as a novel predictor for prognosis and microenvironment remodeling in intrahepatic cholangiocarcinoma》,链接是:https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-024-05238-z
如下所示是的降维聚类分群和命名,很明显的可以看到髓系免疫细胞里面的巨噬细胞和树突细胞的比例是有问题的,而且那些高表达量基因明明是单核细胞的并不是树突细胞的 :
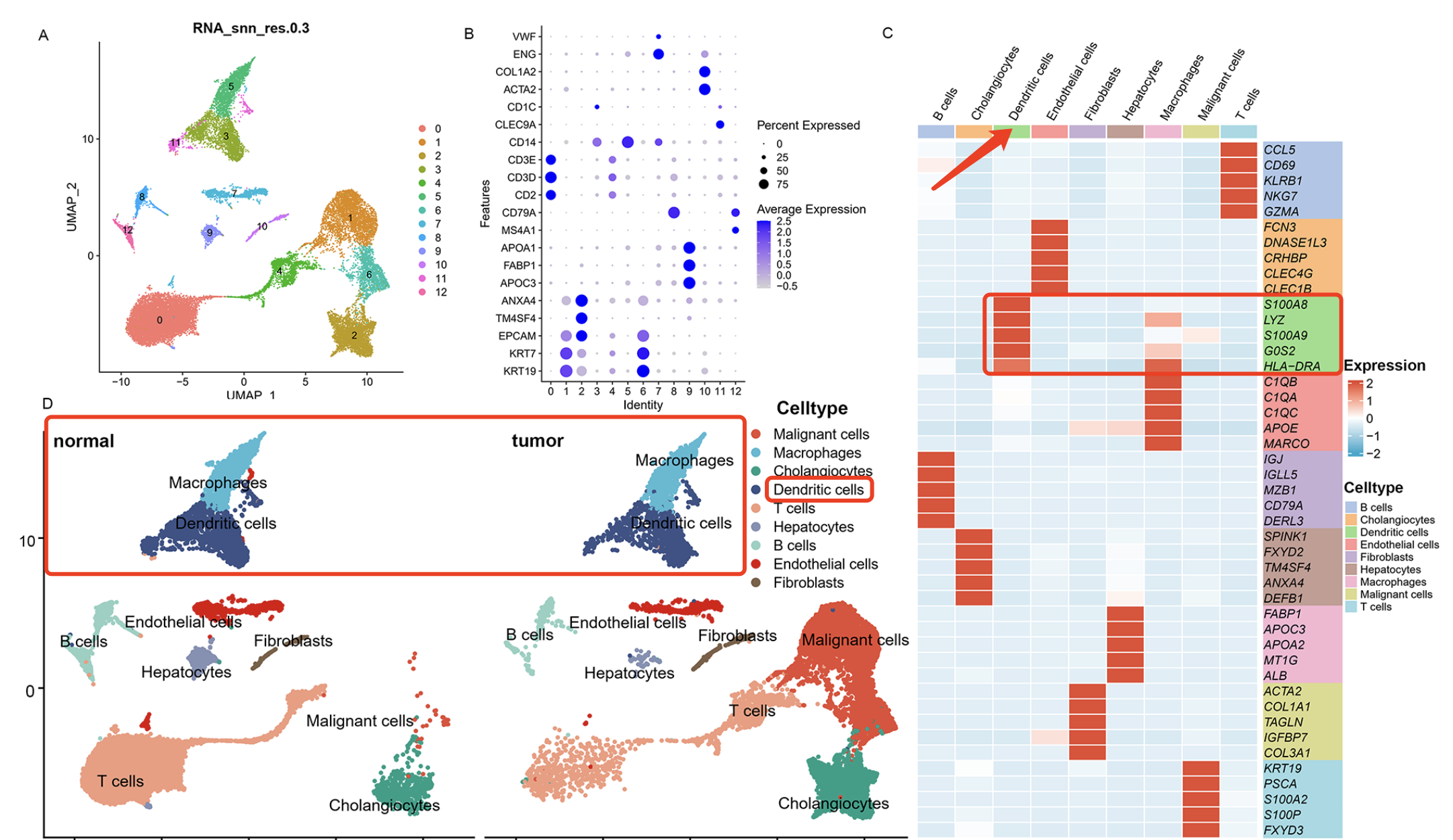
可以看到,研究者们重新理由的是GSE138709这个数据集,很容易下载其表达量矩阵:
15M 10 10 2019 GSM4116579_ICC_18_Adjacent_UMI.csv.gz
16M 10 10 2019 GSM4116580_ICC_18_Tumor_UMI.csv.gz
6.4M 10 10 2019 GSM4116581_ICC_20_Tumor_UMI.csv.gz
8.2M 10 10 2019 GSM4116582_ICC_23_Adjacent_UMI.csv.gz
11M 10 10 2019 GSM4116583_ICC_23_Tumor_UMI.csv.gz
11M 10 11 2019 GSM4116584_ICC_24_Tumor1_UMI.csv.gz
6.9M 10 11 2019 GSM4116585_ICC_24_Tumor2_UMI.csv.gz
3.5M 10 11 2019 GSM4116586_ICC_25_Adjacent_UMI.csv.gz
然后批量读取到Seurat里面即可完成降维聚类分群,只需复制粘贴我的代码(https://pan.baidu.com/s/1bIBG9RciAzDhkTKKA7hEfQ?pwd=y4eh )里面的编号4的文件夹,其中读入的代码是:
dir='GSE138709_RAW'
samples=list.files( dir )
samples
sceList = lapply(samples,function(pro){
# pro=samples[5]
# pro=samples[1]
print(pro)
ct=fread(file.path( dir ,pro),data.table = F)
ct[1:4,1:4]
rownames(ct)=ct[,1]
ct=ct[,-1]
sce=CreateSeuratObject(counts = ct ,
project = gsub('_UMI.csv.gz','',pro))
return(sce)
})
do.call(rbind,lapply(sceList, dim))
samples = gsub('_UMI.csv.gz','',samples)
samples
samples = gsub('^GSM[0-9]*_ICC_','',samples)
samples
names(sceList) = samples
names(sceList)
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ],
add.cell.ids = samples )
names(sce.all@assays$RNA@layers)
sce.all[["RNA"]]$counts
# Alternate accessor function with the same result
LayerData(sce.all, assay = "RNA", layer = "counts")
sce.all <- JoinLayers(sce.all)
dim(sce.all[["RNA"]]$counts )
肿瘤微环境的单细胞亚群比例情况
一般来说,我们拿到了肿瘤相关的单细胞转录组的表达量矩阵后的第一层次降维聚类分群通常是:
- immune (CD45+,PTPRC),
- epithelial/cancer (EpCAM+,EPCAM),
- stromal (CD10+,MME,fibro or CD31+,PECAM1,endo)
参考我前面介绍过 CNS图表复现08—肿瘤单细胞数据第一次分群通用规则,这3大单细胞亚群构成了肿瘤免疫微环境的复杂。绝大部分文章都是抓住免疫细胞亚群进行细分,包括淋巴系(T,B,NK细胞)和髓系(单核,树突,巨噬,粒细胞)的两大类作为第二次细分亚群。但是也有不少文章是抓住stromal 里面的 fibro 和endo进行细分,并且编造生物学故事的。
在肿瘤组织中,单细胞亚群的比例可以因肿瘤类型、分期、患者的个体差异以及肿瘤微环境等多种因素而显著不同。在单细胞RNA测序技术的帮助下,研究人员可以更精确地分析肿瘤微环境中各种细胞亚群的比例和特征。然而,这些比例并不是固定不变的,它们可能随着肿瘤的发展、治疗干预以及患者的免疫状态等因素而变化。具体的细胞亚群比例需要通过实验方法如流式细胞术、免疫组织化学、单细胞RNA测序等来确定,并且每个肿瘤样本都是独特的。
但是,我们人为的处理了接近1000多个不同癌症的单细胞转录组数据集,树突细胞基本上都是占比都低于5%了,甚至都不到1%。前面我们已经介绍了心肝脾肺肾等多个器官的上皮细胞的细分亚群, 以及免疫细胞里面的髓系和B细胞细分亚群:
而且树突细胞细分亚群比较多,是cDC1,cDC2,cDC3,以及pDC,有一些文章里面也把上面的 cDC3 叫做是 mregDC,但是总体上来说,树突细胞不应该是有很大的比例。
数据挖掘很重要,但务必认真
现如今(2024-06-03 )基本上每个疾病都是有成百上千个单细胞转录组数据集公开了,比如肾癌这两天我在咱们的微信交流群就看到下面的3个数据集,实验设计基本上没有区别:
2022-GSE156632-肾癌和癌旁-单细胞
2023-GSE210041-肾癌和癌旁-单细胞
2023-GSE222703-肾癌和癌旁-单细胞
而且都是可以批量读取到Seurat里面即可完成降维聚类分群,只需复制粘贴我的代码(https://pan.baidu.com/s/1bIBG9RciAzDhkTKKA7hEfQ?pwd=y4eh )里面的编号4的文件夹。
但是我们给大家的代码里面的单细胞亚群是需要人工手动命名的,而且我们的代码里面内置了绝大部分亚群的特异性基因,肉眼简单的检查一下即可 :
myeloids_markers_list3 = list(
Mac=c("C1QA","C1QB","C1QC","SELENOP","RNASE1","DAB2","LGMN","PLTP","MAF","SLCO2B1"),
mono=c("VCAN","FCN1","CD300E","S100A12","EREG","APOBEC3A","STXBP2","ASGR1","CCR2","NRG1"),
neutrophils = c("FCGR3B","CXCR2","SLC25A37","G0S2","CXCR1","ADGRG3","PROK2","STEAP4","CMTM2" ),
pDC = c("GZMB","SCT","CLIC3","LRRC26","LILRA4","PACSIN1","CLEC4C","MAP1A","PTCRA","C12orf75"),
DC1 = c("CLEC9A","XCR1","CLNK","CADM1","ENPP1","SNX22","NCALD","DBN1","HLA-DOB","PPY"),
DC2= c( "CD1C","FCER1A","CD1E","AL138899.1","CD2","GPAT3","CCND2","ENHO","PKIB","CD1B"),
DC3 = c("HMSD","ANKRD33B","LAD1","CCR7","LAMP3","CCL19","CCL22","INSM1","TNNT2","TUBB2B")
)
生命科学领域正在迅速发展,其中公共数据集的利用变得日益重要,原因包括:
-
数据共享:科学界越来越重视数据共享,这有助于加速研究进程,促进知识的积累和创新。
-
大规模数据分析:现代生命科学研究常常需要分析大规模的数据集,这些数据集往往超出了单个实验室的处理能力。
-
成本效益:利用现有的公共数据集可以节省研究成本,避免重复工作。
-
国际合作:公共数据集促进了国际间的科研合作,不同国家和地区的研究人员可以共同分析和解释数据。
-
数据标准化:公共数据集通常遵循标准化的格式和协议,这有助于不同研究之间的比较和整合。
然而,准确性在利用公共数据集时至关重要,以下是一些关键点:
-
数据质量:公共数据集的质量参差不齐,使用前需要进行严格的质量控制。
-
数据注释:数据集中的元数据(如样本信息、实验条件、测序平台等)需要准确无误,以便正确解释数据。
-
数据完整性:确保数据集的完整性,避免因数据缺失或损坏而导致的错误结论。
-
数据可追溯性:研究者应能够追溯数据的来源和处理历史,以验证数据的可靠性。
-
统计分析:使用适当的统计方法来分析数据,避免统计错误。
-
数据解释:对数据的解释需要谨慎,考虑到可能的偏差和局限性。
-
更新和维护:随着时间的推移,数据集可能需要更新和维护,以确保其反映最新的科学发现。
-
伦理和隐私:在使用涉及个人信息的数据集时,需要遵守伦理和隐私保护的规定。
为了确保使用公共数据集的准确性,研究人员通常会:
- 对数据进行预处理和清洗,以提高数据质量。
- 采用严格的统计方法和生物信息学工具。
- 与数据集的原始作者或维护者合作,以确保正确理解和使用数据。
- 发表研究时,详细报告数据来源、处理方法和分析过程。
总之,公共数据集在生命科学中扮演着越来越重要的角色,但确保数据的准确性和可靠性是得出有效科学结论的关键。
如果你不确定自己的数据分析是否正确或者说合理
可以试试看我们的数据分析顾问服务哈, 否则很容易被diss,参考:单细胞,单细胞,我要diss你!
或者起码在正规机构系统性学习一下,而不是网络上面随便抄几个代码或者参加一个两天的速成班。。。
至于正规机构系统性学习,我 推荐《生信技能树》团队的