这些年陆陆续续处理了一两千个单细胞转录组公共数据集了,总是会碰到一些缺胳膊断腿的,比如:GSE233447
GSM7428203 Uterine_neuroendocrine [OE180716630-01]
GSM7428204 Uterine_neuroendocrine [OE180716630-02]
GSM7428205 Uterine_neuroendocrine [OE180716630-03]
GSM7428206 Uterine_neuroendocrine [OE180716630-04]
首先从这个GEO页面的介绍看,还以为是4个样品的10x单细胞转录组,但是实际上是单个样品: a specimen obtained from a patient diagnosed with small cell endometrial neuroendocrine carcinoma (SCNEC) based on pathology. 然后降维聚类分群和命名后,如下所示:
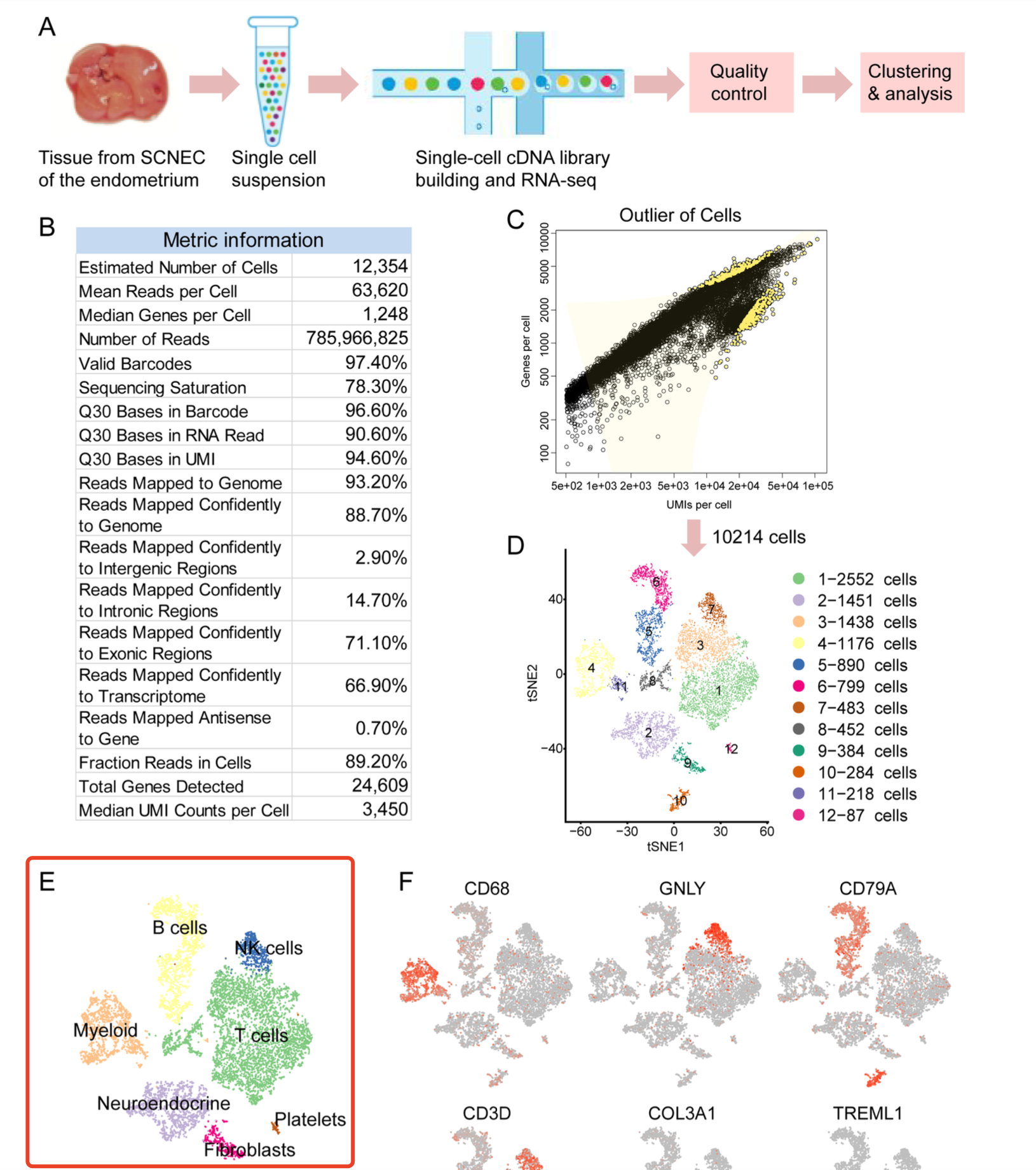
但是研究者公开的就是70多M的GSE233447_matrix.mtx.gz文件而已,确实是可以读取:
ct=Matrix::readMM('GSE233447_matrix.mtx.gz' )
ct[1:4,1:4]
dim(ct)
## 如下所示
4 x 4 sparse Matrix of class "dgTMatrix"
[1,] . . . .
[2,] . . . .
[3,] . . . .
[4,] . . . .
> dim(ct)
[1] 33538 12354
很明显,是 12354个单细胞,然后有33538个基因,但是作者并没有给出来细胞的id和基因的symbol,这样的数据就好尴尬!
我这里这一个狡猾的操作:
rownames(ct)=paste0('r',1:nrow(ct))
colnames(ct)=paste0('c',1:ncol(ct))
然后可以跑降维聚类分群啦, 但是因为没有基因名字,所以是不可能针对不同单细胞亚群给出来合理的生物学名字;
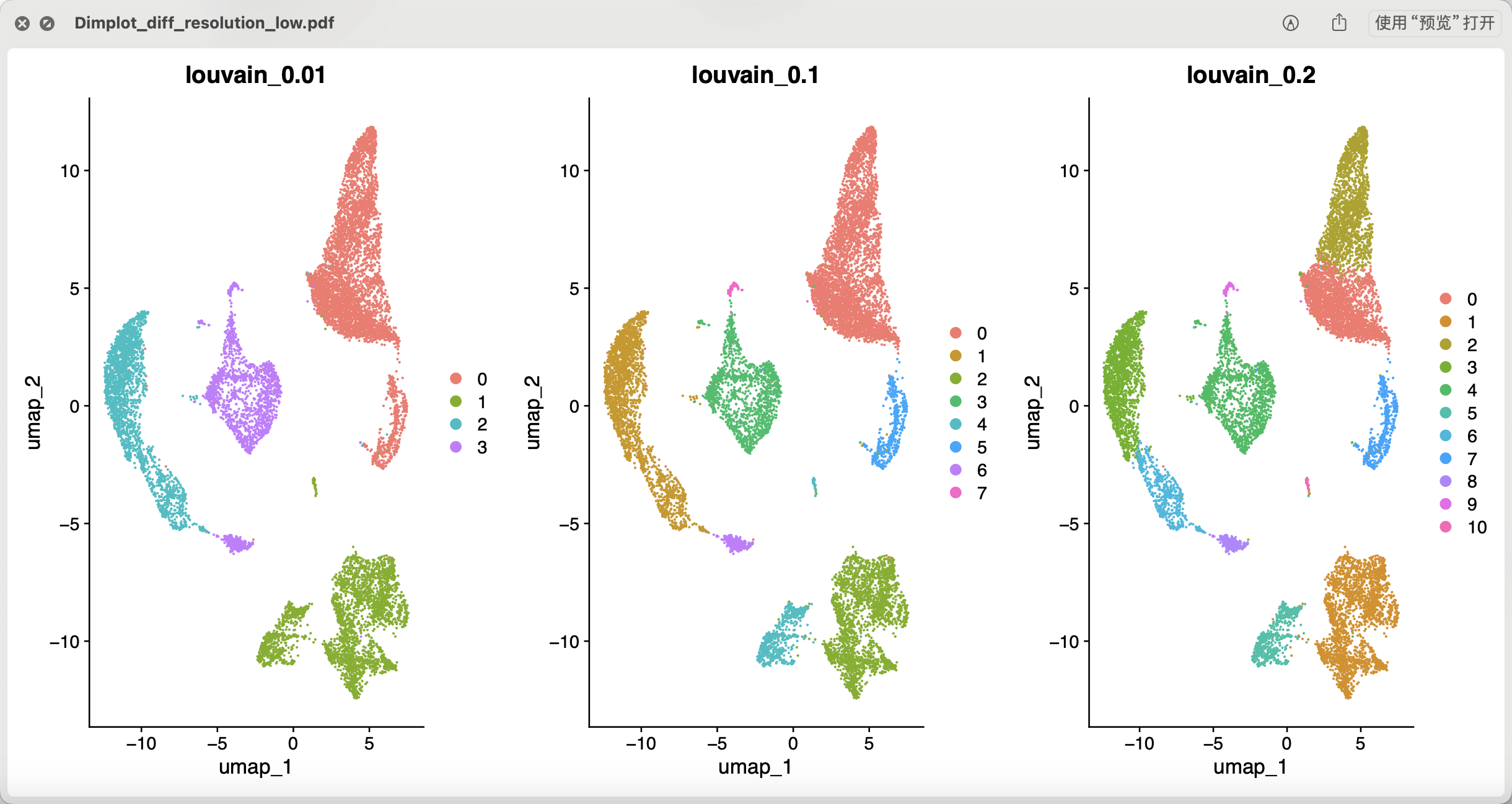
但是因为作者给出来了单细胞亚群的细胞数量,所以我们勉强是可以对应一下:
> as.data.frame(table(sce.all.int$RNA_snn_res.0.2) )
Var1 Freq
1 0 2621
2 1 2332
3 2 1968
4 3 1817
5 4 1539
6 5 608
7 6 562
8 7 491
9 8 271
10 9 95
11 10 50
需要强大的逻辑推理能力,我感觉没必要。。。
还不如直接去找作者要对应的基因名字文件即可,是同济大学医学院附属上海肺科医院的检验医学系的Yin Liu ,他也有邮箱给出来( liuyin@tongji.edu.cn),感兴趣的也可以读一下他的文章哈: Single-Cell Transcriptome Analysis of Small Cell Neuroendocrine Carcinoma of the Endometrium Reveals ISL1 as a Potential Biomarker for Diagnosis and Treatment. Front Biosci (Landmark Ed) 2024 Mar 13;29(3):100. PMID: 38538277
学徒作业
上面的GSE233447这个数据集其实有对应的fastq文件,在:https://www.ncbi.nlm.nih.gov/bioproject/PRJNA976283,大家可以使用服务器对它走上游流程。(而且我也是比较好奇,为什么明明是一个样品,会拆分成为了4个 )
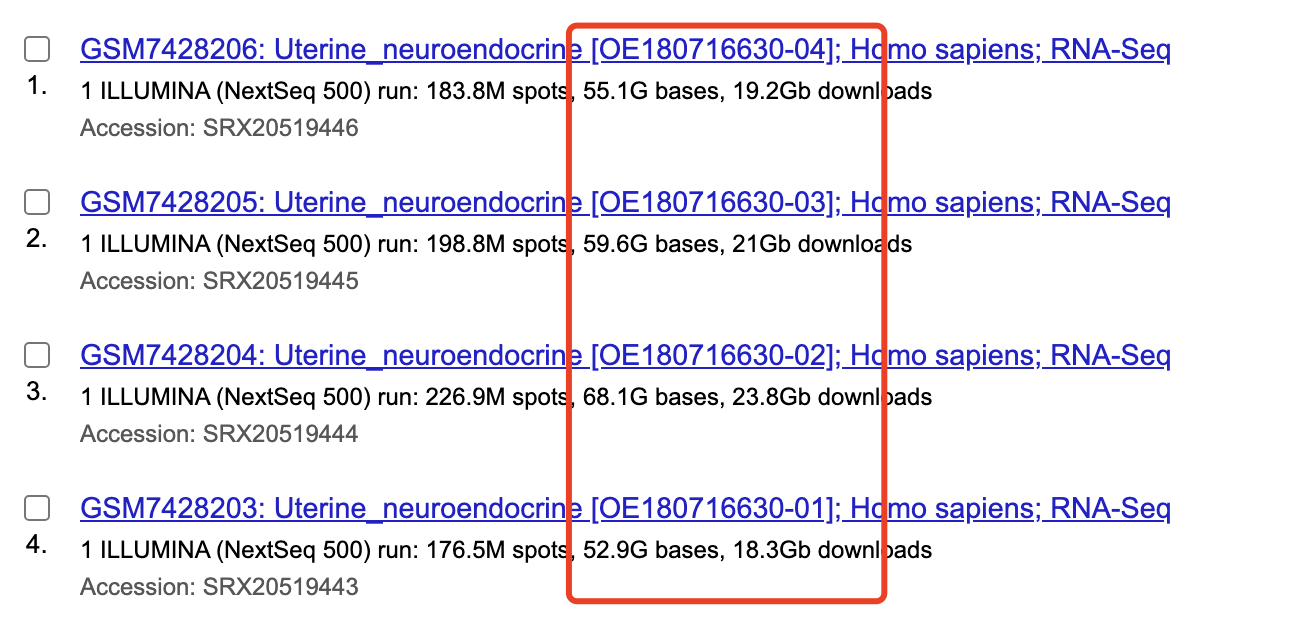
正常走cellranger的定量流程即可,代码我已经是多次分享了。参考:
- 10X单细胞转录组原始测序数据的Cell Ranger流程(仅需800元)
- 10X的单细胞转录组原始数据也可以在EBI下载
- 一个10x单细胞转录组项目从fastq到细胞亚群
- 一文打通单细胞上游:从软件部署到上游分析
- PRJNA713302这个10x单细胞fastq实战
- 一次曲折且昂贵的单细胞公共数据获取与上游处理
- 只能下载bam文件的10x单细胞转录组项目数据处理
- 不知道10x单细胞转录组样品和fastq文件的对应关系
- 10X单细胞转录组测序数据的 SRA转fastq踩坑那些事
- 10x的单细胞转录组fastq文件的R1和R2不能弄混哦
差不多几个小时就可以完成全部的样品的cellranger的定量流程。