RSeQC对 RNA-seq数据质控
一:下载安装该软件
wget http://sourceforge.net/projects/rseqc/files/RSeQC-2.5.tar.gz/download
tar zxvf RSeQC.tar.gz
cd RSeQC-2.5/
进入目录查看文件内容如下,看到了setup.py,所以知道这个是一个python的包,需要安装到python的库下面。
我没有root权限,所以需要把这个软件安装到自己的目录下,同时会新建一个python的库路径,需要添加到python的库搜索路径
python setup.py install --root=/home/jmzeng/my-bin
安装完毕后,这个python模块在/home/jmzeng/my-bin/usr/local/lib/python2.7/dist-packages/里面
安装完毕后,/home/jmzeng/my-bin/usr/local/bin 这个目录下面会多一堆的程序,就是我们这个 RSeQC软件包的东西啦
但是这个目录并不在我的PATH里面,需要添加
而且这样安装后面会报错,因为很多模块都不在路径里面~!~~~~当然把我自己的安装的python库添加到系统python的环境变量里面
所以python setup.py install --user 这样才是正确的
安装好后,所有这个包带有的python程序都在/home/jmzeng/.local/bin这个目录里面~!!!
非常好用,也不会出现下面的错误了
二:准备数据
准备数据
该软件有提供一些测试数据,但是存放在drive.google.com里面,国内访问不是很容易,我们就根据要求自己创造一些吧,BED文件个SAM文件还有参考基因组的染色体长度信息文件和原始的reads的fasta格式文件。我这里是取的一个RNA-seq的测序fastq格式文件用tophat比对后的bam文件来做示范。

三:运行命令 && 四:输出文件解读
因为这是一个python软件套装,里面至少含有十几个工具,我这里简单介绍几个常用的,其余的待有需要再去看看。
1)bam_stat.py
比对片段检查(QC 失败、unique mapped、splice mapped、mapped 到合适对的 reads 等)
例子: bam_stat.py -i Pairend_nonStrandSpecific_36mer_Human_hg19.bam
我的命令python /home/jmzeng/my-bin/usr/local/bin/bam_stat.py -i accepted_hits.bam

这是一个很变态的bug,ImportError: No module named qcmodule。是python这个垃圾语言造成的,因为我没有服务器root权限,所以我的python包只能安装到自己的目录,但是我调用了服务器的python命令,它却不识别我的python库,变态!
查了一些资料,原来需要把我自己的安装的python库添加到系统python的环境变量里面
export PYTHONPATH=/home/jmzeng/my-bin/usr/local/lib/python2.7/dist-packages/:$PYTHONPATH
然后几分钟就出结果啦
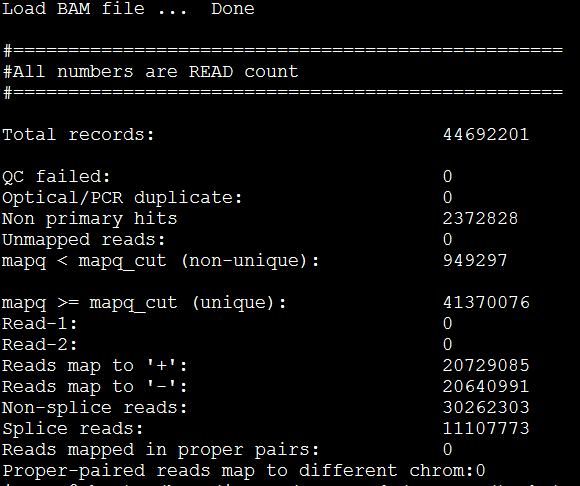
因为我这个是单端测序,所以没有reads1和2的区别。
2)clipping_profile.py
This program is used to estimate clipping profile of RNA-seq reads from BAM or SAM file.Note that to use this funciton, CIGAR strings within SAM/BAM file should have ‘S’ operation(This means your reads aligner should support clipped mapping).
例子 : clipping_profile.py -i Pairend_StrandSpecific_51mer_Human_hg19.bam -o output
我的命令:python /home/jmzeng/my-bin/usr/local/bin/clipping_profile.py -i accepted_hits.bam -o output
然后就等了几分钟
Load BAM file ... Done
No clipped reads found!
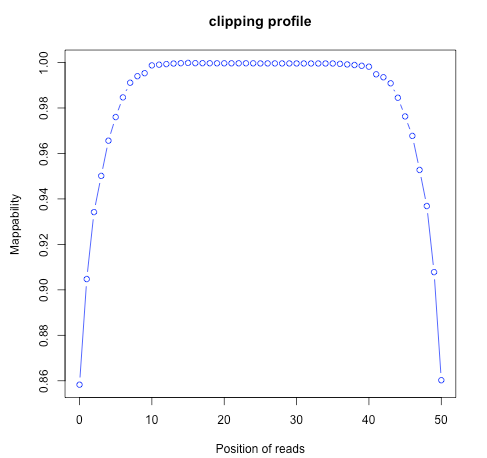
程序报错很奇怪,输出了一个xls的表格,但是里面只有表头没有数据,所以我认为应该是数据出了什么问题吧
理论上是应该要出一个如下所示的图的
3)mismatch_profile.py
Calculate the distributions of the mismatches across reads. Note that the “MD” tag must exist in the BAM file.
理论上也是应该出一个图
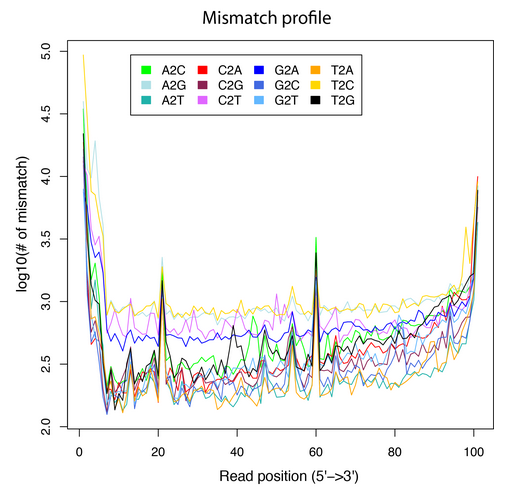
4)insertion_profile.py
Calculate the distributions of (short) insertions across reads.
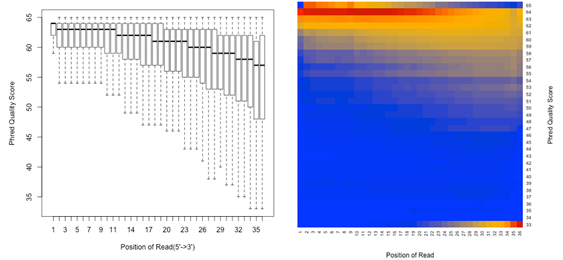
5)read_quality.py
这个就是fastqc的质量检测啦,这个看起来挺有用的,我也画了一个图
python /home/jmzeng/my-bin/usr/local/bin/read_quality.py -i accepted_hits.bam -o output
输出两个pdf图片文件
6)bam2wig.py
这是一个格式转换工具,把BAM文件转为wiggle格式的文件
还可以把我们转好的wiggle格式文件通过UCSC wigToBigWig 这个工具转为bigwig format
为了使用这个脚本,我们的bam文件必须是排序好了的,并且还有索引。# sort and index BAM files
samtools sort -m 1000000000 input.bam input.sorted.bam
samtools index input.sorted.bam
7)bam2fq.py
也是一个格式转换工具,可以把bam转为fastq格式,没啥子意思,一大堆的软件都附带这个工具。
这里面的程序太多了,每个都可以出图,懒得一个个试用并且翻译了,自己看吧http://rseqc.sourceforge.net
