de novo变异寻找这个也属于snp-calling的一部分,但是有点不同的就是该软件考虑了一家三口的测序文件,找de novo突变
软件有介绍这个功能:http://varscan.sourceforge.net/trio-calling-de-novo-mutations.html
而且还专门有一篇文章讲ASD和autism与de novo变异的关系,但是文章不清不楚的,没什么意思
Trio Calling for de novo Mutations
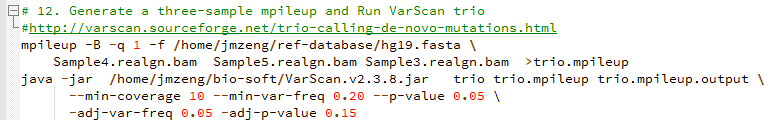
Min coverage: 10
Min reads2: 4
Min var freq: 0.2
Min avg qual: 15
P-value thresh: 0.05
Adj. min reads2: 2
Adj. var freq: 0.05
Adj. p-value: 0.15
Reading input from trio.filter.mpileup
1371416525 bases in pileup file (137M的序列)
83123183 met the coverage requirement of 10 (其中有83M的测序深度大于10X)
145104 variant positions (132268 SNP, 12836 indel) (共发现15.5万的变异位点)
4403 were failed by the strand-filter
139153 variant positions reported (126762 SNP, 12391 indel)
502 de novo mutations reported (376 SNP, 126 indel) (真正属于 de novo mutations只有502个)
1734 initially DeNovo were re-called Germline
12 initially DeNovo were re-called MIE
3 initially DeNovo were re-called MultAlleles
522 initially MIE were re-called Germline
1 initially MIE were re-called MultAlleles
3851 initially Untransmitted were re-called Germline
然后我看了看输出的文件trio.mpileup.output.snp.vcf
软件是这样解释的:The output of the trio subcommand is a single VCF in which all variants are classified as germline (transmitted or untransmitted), de novo, or MIE.
- FILTER - mendelError if MIE, otherwise PASS
- STATUS - 1=untransmitted, 2=transmitted, 3=denovo, 4=MIE
- DENOVO - if present, indicates a high-confidence de novo mutation call
里面的信息量好还是不清楚
我用annovar注释了一下
/home/jmzeng/bio-soft/annovar/convert2annovar.pl -format vcf4 trio.mpileup.output.snp.vcf > trio.snp.annovar
/home/jmzeng/bio-soft/annovar/annotate_variation.pl -buildver hg19 --geneanno --outfile trio.snp.anno trio.snp.annovar /home/jmzeng/bio-soft/annovar/humandb
结果是:
A total of 132268 locus in VCF file passed QC threshold, representing 132809 SNPs (86633 transitions and 46176 transversions) and 3 indels/substitutions
可以看到最后被注释到外显子上面的突变有两万多个
23794 284345 3123333 trio.snp.anno.exonic_variant_function
这个应该是非常有意义的,但是我还没学会后面的分析。只能先做到这里了