limma真不愧是最流行的差异分析包,十多年过去了,一直是芯片数据处理的好帮手。
现在又可以支持RNA-seq数据,我赶紧试用了一下!
我下面只讲用法,大家看代码就明白了!
##
library(limma)
library(pasilla)
data(pasillaGenes)
exprSet=counts(pasillaGenes)
group_list=pasillaGenes$condition
## 只需自己构造好表达矩阵exprSet和分因子即可group_list,一般只分成两组!!!
##一般是自己读取RNA-seq的基因的reads的counts数进行分析,##请不要用RPKM等经过了normlization的表达矩阵来分析。
suppressMessages(library(limma))
design <- model.matrix(~factor(group_list))
colnames(design)=levels(factor(group_list))
rownames(design)=colnames(exprSet)
v <- voom(exprSet,design,normalize="quantile") ##这个是重点
## 到这里就跟limma本身的用法一样了!
fit <- lmFit(v,design)
fit2 <- eBayes(fit)
tempOutput = topTable(fit2, coef=2, n=Inf)
DEG_voom = na.omit(tempOutput)
head(DEG_voom)
它也是用了一种统计方法,把RNA-seq的基因的reads的counts数进行了normlization
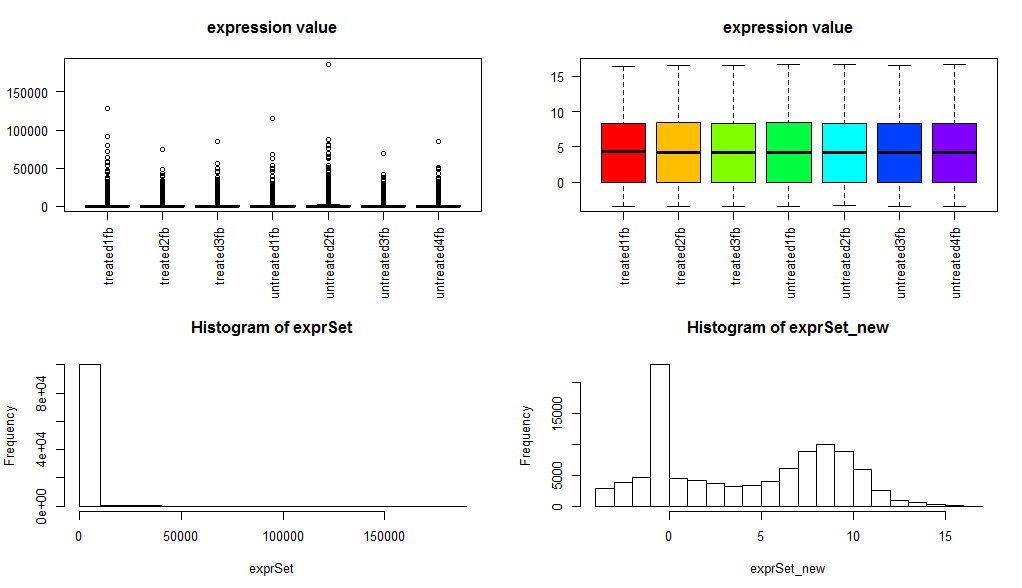
看这个图就知道了,它把本来应该是数据离散程度非常大的RNA-seq的基因的reads的counts矩阵经过normlization后变成了类似于芯片表达数据的表达矩阵,然后其实可以直接用T检验来找差异基因了!
但是,如果你的分组不只是两个,就复杂了,你需要再仔细研读说明书,甚至你可能需要咨询实验设计人员或者统计人员!