CHIP-seq测序的本质还是目标片段捕获测序,跟WES不同的是,它不是通过固定的芯片探针来固定的捕获基因组上面特定序列,而是根据你选择的IP不同,你细胞或者机体状态不同,捕获到的序列差异很大!而我们研究的重点,就是捕获到的差异。而我们对CHIP-seq测序数据寻找peaks的本质就是得到所有测序数据比对在全基因组之后在正个基因组上面的测序深度里面寻找比较突出的。比如对WES数据来说,各个外显子,或者外显子的5端到3端,理论上测序深度应该是一致的,都是50X~200X,画一个测序深度曲线,应该是近似于一条直线。对我们的CHIP-seq测序数据来说,在所捕获的区域上面,理论上测序深度是绝对不一样的,应该是近似于一个山峰。而那些覆盖度高的地方,山顶,就是我们的IP所结合的热点,也就是我们想要找的peaks,在IGV里面看到大致是下面这样:
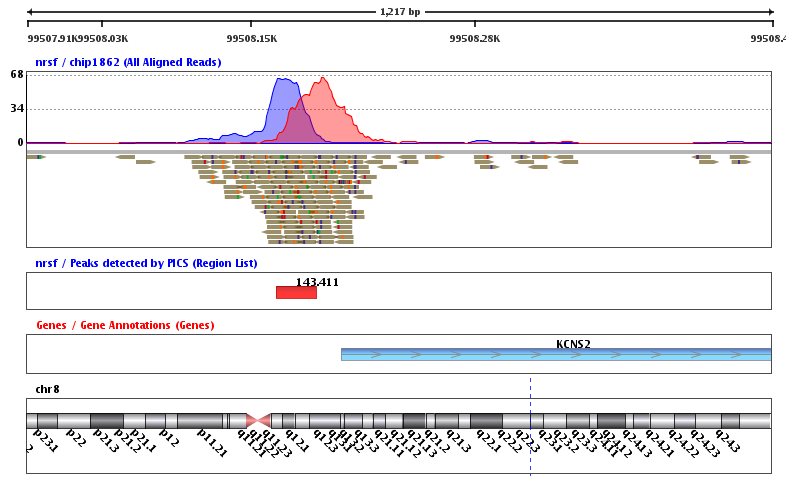
可以看到测序的reads分布是绝对的不均匀的!我们通常说的CHIP-seq测序的IP,可以是各个组蛋白的各个修饰位点对应的抗体,或者是各种转录因子的抗体,等等
如何定义热点呢?通俗地讲,热点是这样一些位置,这些位置多次被测得的read所覆盖(我们测的是一个细胞群体,read出现次数多,说明该位置被TF结合的几率大)。那么,read数达到多少才叫多?这就要用到统计检验喽。假设TF在基因组上的分布是没有任何规律的,那么,测序得到的read在基因组上的分布也必然是随机的,某个碱基上覆盖的read的数目应该服从二项分布。
具体统计学原理直接看原创吧:http://www.plob.org/2014/05/08/7227.html
为了达到作者文献里面的结果,我换了8个软件:MACS2/HOMER/SICERpy/PePr/SWEMBL/SISSRs/BayesPeak/PeakRanger,我这里就不一一介绍peaks caller软件的安装以及使用了,因为MACS2是最常用的,我就简单贴一下我关于MACS2的学习代码:
## step6 : peak calling
### step6.1: with MACS2
## 我先看了看说明书:
macs2 callpeak -t TF_1.bam -c Input.bam -n mypeaks
We used the following options:
-t: This is the only required parameter for MACS, refers to the name of the file with the ChIP-seq data
-c: The control or mock data file
-n: The name string of the experiment
MAC2 creates 4 files (mypeaks peaks.narrowPeak, mypeaks summits.bed, mypeaks peaks.xls and mypeaks model.r)
# MACS首先的工作是要确定一个模型,这个模型最关键的参数就是峰宽d。这个d就是bw(band width),而它的一半就是shiftsize。### 然后根据文章确定了下载的测序数据的分类
GSM1278641 Xu_MUT_rep1_BAF155_MUT SRR1042593
GSM1278642 Xu_MUT_rep1_Input SRR1042594
GSM1278643 Xu_MUT_rep2_BAF155_MUT SRR1042595
GSM1278644 Xu_MUT_rep2_Input SRR1042596
GSM1278645 Xu_WT_rep1_BAF155 SRR1042597
GSM1278646 Xu_WT_rep1_Input SRR1042598
GSM1278647 Xu_WT_rep2_BAF155 SRR1042599
GSM1278648 Xu_WT_rep2_Input SRR1042600
## 这里有个很奇怪的问题,input的测序数据居然比IP的测序数据多???
848M Jun 28 14:31 SRR1042593.bam
2.7G Jun 28 14:52 SRR1042594.bam
716M Jun 28 14:58 SRR1042595.bam
2.9G Jun 28 15:20 SRR1042596.bam
1.1G Jun 28 15:28 SRR1042597.bam
2.6G Jun 28 15:48 SRR1042598.bam
1.2G Jun 28 15:58 SRR1042599.bam
3.5G Jun 28 16:26 SRR1042600.bam
## 我没有想明白为什么
## http://www2.uef.fi/documents/1698400/2466431/Macs2/f4d12870-34f9-43ef-bf0d-f5d087267602
## http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3120977/我首先用的是下面这些代码
nohup time ~/.local/bin/macs2 callpeak -c SRR1042594.bam -t SRR1042593.bam -f BAM -B -g hs -n Xu_MUT_rep1 2>Xu_MUT_rep1.masc2.log &
nohup time ~/.local/bin/macs2 callpeak -c SRR1042596.bam -t SRR1042595.bam -f BAM -B -g hs -n Xu_MUT_rep2 2>Xu_MUT_rep2.masc2.log &
nohup time ~/.local/bin/macs2 callpeak -c SRR1042598.bam -t SRR1042597.bam -f BAM -B -g hs -n Xu_WT_rep1 2>Xu_WT_rep1.masc2.log &
nohup time ~/.local/bin/macs2 callpeak -c SRR1042600.bam -t SRR1042599.bam -f BAM -B -g hs -n Xu_WT_rep2 2>Xu_WT_rep2.masc2.log &
得到的peaks少的可怜,我第一次检查,以为是因为自己没有sort 比对的bam文件导致
## forget to sort the bam files:
## 首先把bam文件sort好,构建了inde,然后继续运行!
nohup time ~/.local/bin/macs2 callpeak -c SRR1042594.sorted.bam -t SRR1042593.sorted.bam -f BAM -B -g hs -n Xu_MUT_rep1 2>Xu_MUT_rep1.masc2.log &
nohup time ~/.local/bin/macs2 callpeak -c SRR1042596.sorted.bam -t SRR1042595.sorted.bam -f BAM -B -g hs -n Xu_MUT_rep2 2>Xu_MUT_rep2.masc2.log &
nohup time ~/.local/bin/macs2 callpeak -c SRR1042598.sorted.bam -t SRR1042597.sorted.bam -f BAM -B -g hs -n Xu_WT_rep1 2>Xu_WT_rep1.masc2.log &
nohup time ~/.local/bin/macs2 callpeak -c SRR1042600.sorted.bam -t SRR1042599.sorted.bam -f BAM -B -g hs -n Xu_WT_rep2 2>Xu_WT_rep2.masc2.log &
##此时得到peaks跟上面为sort的bam文件得到的peaks一模一样,看来不是这个原因##然后我怀疑是不是作者上传数据的时候把input和IP标记反了,所以我认为的调整过来## Then change the control and treatment
nohup time ~/.local/bin/macs2 callpeak -t SRR1042594.sorted.bam -c SRR1042593.sorted.bam -f BAM -B -g hs -n Xu_MUT_rep1 2>Xu_MUT_rep1.masc2.log &
nohup time ~/.local/bin/macs2 callpeak -t SRR1042596.sorted.bam -c SRR1042595.sorted.bam -f BAM -B -g hs -n Xu_MUT_rep2 2>Xu_MUT_rep2.masc2.log &
nohup time ~/.local/bin/macs2 callpeak -t SRR1042598.sorted.bam -c SRR1042597.sorted.bam -f BAM -B -g hs -n Xu_WT_rep1 2>Xu_WT_rep1.masc2.log &
nohup time ~/.local/bin/macs2 callpeak -t SRR1042600.sorted.bam -c SRR1042599.sorted.bam -f BAM -B -g hs -n Xu_WT_rep2 2>Xu_WT_rep2.masc2.log &##结果,压根就没有peaks了!!!!看了作者并没有搞错
##接下来我怀疑是自己用samtools view -bhS -q 30 处理了sam文件,这个标准太严格了!!#### then just use the sam files.
nohup time ~/.local/bin/macs2 callpeak -c SRR1042594.sam -t SRR1042593.sam -f SAM -B -g hs -n Xu_MUT_rep1 2>Xu_MUT_rep1.masc2.log &
nohup time ~/.local/bin/macs2 callpeak -c SRR1042596.sam -t SRR1042595.sam -f SAM -B -g hs -n Xu_MUT_rep2 2>Xu_MUT_rep2.masc2.log &
nohup time ~/.local/bin/macs2 callpeak -c SRR1042598.sam -t SRR1042597.sam -f SAM -B -g hs -n Xu_WT_rep1 2>Xu_WT_rep1.masc2.log &
nohup time ~/.local/bin/macs2 callpeak -c SRR1042600.sam -t SRR1042599.sam -f SAM -B -g hs -n Xu_WT_rep2 2>Xu_WT_rep2.masc2.log &
## 也没有多几个peaks,最后我只能想到是我的p值太严格了
## then chang the criteria for p values :https://github.com/taoliu/MACS/
nohup time ~/.local/bin/macs2 callpeak -c SRR1042594.sam -t SRR1042593.sam -f SAM -p 0.01 -g hs -n Xu_MUT_rep1 2>Xu_MUT_rep1.masc2.log &
nohup time ~/.local/bin/macs2 callpeak -c SRR1042596.sam -t SRR1042595.sam -f SAM -p 0.01 -g hs -n Xu_MUT_rep2 2>Xu_MUT_rep2.masc2.log &
nohup time ~/.local/bin/macs2 callpeak -c SRR1042598.sam -t SRR1042597.sam -f SAM -p 0.01 -g hs -n Xu_WT_rep1 2>Xu_WT_rep1.masc2.log &
nohup time ~/.local/bin/macs2 callpeak -c SRR1042600.sam -t SRR1042599.sam -f SAM -p 0.01 -g hs -n Xu_WT_rep2 2>Xu_WT_rep2.masc2.log &
##我大大减小了P值的标准,结果是输出一大堆的peaks
18919 Xu_MUT_rep1_peaks.xls
36277 Xu_MUT_rep2_peaks.xls
32494 Xu_WT_rep1_peaks.xls
56080 Xu_WT_rep2_peaks.xls
问题是这些peaks根本就都是假阳性!!!
我手动的check了几个之前严格过滤条件下的peaks,的确可以看到测序深度是两个山峰形状的曲线
## check some peaks 手动的 ## chr1 121484235 121485608
## masc results :
samtools depth -r chr10:42385331-42385599 SRR1042593.sorted.bam
samtools depth -r chr10:42385331-42385599 SRR1042594.sorted.bam
samtools depth -r chr20:45810382-45810662 SRR1042593.sorted.bam
samtools depth -r chr20:45810382-45810662 SRR1042594.sorted.bam
##我也check了paper里面得到的peak,但是在我的比对文件里面,肉眼看起来根本不像,所以我很纠结~~~~
paper results:
chr20 45796362 46384917
chr1 121482722 121485861
samtools depth -r chr1:121482722-121485861 SRR1042593.sorted.bam
samtools depth -r chr1:121482722-121485861 SRR1042594.sorted.bam
samtools depth -r chr20:45796362-46384917 SRR1042593.sorted.bam
samtools depth -r chr20:45796362-46384917 SRR1042594.sorted.bam