对于CHIP-seq数据处理完全是自学的,所以有很多细节得慢慢学习回来,这次记录的就是当我们把测序仪的fastq数据比对到参考基因组之后,应该对比对的结果文件做什么样的处理,然后去给peaks caller软件拿来call peaks呢?我看过博客 提到只保留比对质量值大于30的,也看过博客提到只保留unique比对的reads,我这里拿一篇公共数据测试了一下它们的区别!数据描述如下:
| GSM1916974 | H3K27ac ChIP-seq |
SRR2774675 |
| GSM1916975 | input DNA |
SRR2774676 |
首先在SRA数据库下载 SRR2774675.sra 和 SRR2774676.sra
应用我github的流程很快就可以对比对,我把两种方法处理的比对结果都拿去call peaks,然后得到了,两个peaks文件。
39709 highQuaily_peaks.bed
39709 highQuaily_summits.bed
39709 highQuaily_summits.bed
可以看到两次结果得到的peaks条数并没有显著区别,我们简单看看前几行!
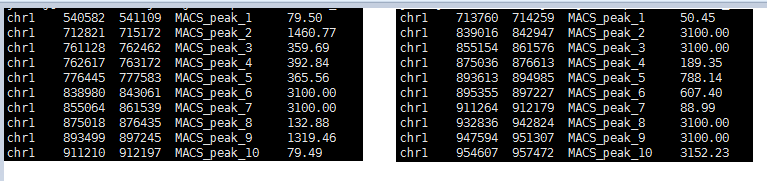
其实用bedtools就可以看看左右两边的文件的交集情况,但是我这里选用了ChIPpeakAnno这个R包集成好的函数,直接得到结果即可!
ChIPpeakAnno 包直接看说明书吧,我这里贴出代码:
library(ChIPpeakAnno)
highPeak <- readPeakFile( 'highQuaily_peaks.bed' )
uniquePeak <- readPeakFile( 'unique_peaks.bed' )
ol <- findOverlapsOfPeaks(highPeak, uniquePeak)
png('overlapVenn.png')
makeVennDiagram(ol)
dev.off()
然后打开画好的韦恩图:
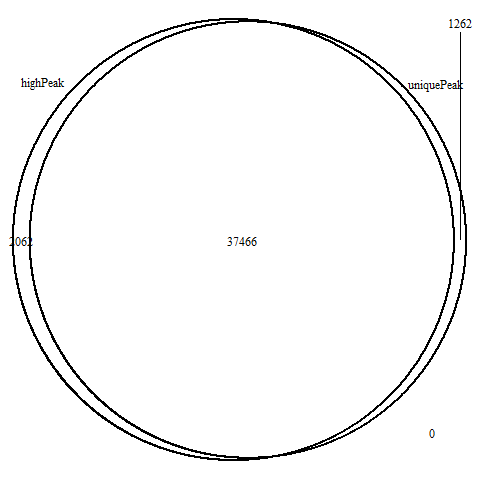
可以看到这两种情况得到的结果几乎没有区别,如果大家感兴趣可以自己看看它们那些独特的peaks到底是什么原因!
结论就是,说明CHIP-seq数据分析的时候,call peaks那个步骤,只保留比对质量值大于30的,或者只保留unique比对的reads,从数据处理的角度来讲差别不大,主要看你具体实验意义。