因为一直拿到的是公司给的特别好的数据,所以没太关注质控这个问题,最近拿到了raw data,才发现其实里面的门道挺多的。前面都是用cutadapt这个python软件来去除接头的,但是它有一个弊端,需要自己指定接头文件。正好朋友推荐了trimmomatic,是java软件,所以直接Google找到其官网,然后下载二进制版本解压即可使用!
反正对我的illumina测序数据来说,直接用它就可以把raw data 变成 clean data啦!

这个软件设计就是为了illumina的测序数据的,因为它自带的adaptor文件有限,上图可以看到!而且一般只去除TruSeq Universal Adapter 这个接头,运行的时候,不报错才算是成功的!
Paired End:
java -jar trimmomatic-0.35.jar PE -phred33 input_forward.fq.gz input_reverse.fq.gz output_forward_paired.fq.gz output_forward_unpaired.fq.gz output_reverse_paired.fq.gz output_reverse_unpaired.fq.gz ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36 ## 所以只需要把参数放对位置即可!
This will perform the following:
- Remove adapters (ILLUMINACLIP:TruSeq3-PE.fa:2:30:10)
- Remove leading low quality or N bases (below quality 3) (LEADING:3)
- Remove trailing low quality or N bases (below quality 3) (TRAILING:3)
- Scan the read with a 4-base wide sliding window, cutting when the average quality per base drops below 15 (SLIDINGWINDOW:4:15)
- Drop reads below the 36 bases long (MINLEN:36)
一般就使用这个默认参数就好啦,处理的时间会有一点慢,我取了10个线程也得十几分钟才搞定2G的fq.gz压缩格式的测序文件,文件的log日志如下:
TrimmomaticPE: Started with arguments:
-threads 10 -phred33 -trimlog tmp.log CHG006373_R1.fastq.gz CHG006373_R2.fastq.gz output_forward_paired.fq.gz output_forward_unpaired.fq.gz output_reverse_paired.fq.gz output_reverse_unpaired.fq.gz ILLUMINACLIP:/home/jmzeng//biosoft/trimmomatic/Trimmomatic-0.36/adapters/TruSeq3-PE.fa:2:30:10 LEADING:10 TRAILING:20 SLIDINGWINDOW:4:25 MINLEN:36
Using PrefixPair: 'TACACTCTTTCCCTACACGACGCTCTTCCGATCT' and 'GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT'
ILLUMINACLIP: Using 1 prefix pairs, 0 forward/reverse sequences, 0 forward only sequences, 0 reverse only sequences
Input Read Pairs: 21427010 Both Surviving: 14507723 (67.71%) Forward Only Surviving: 5297811 (24.72%) Reverse Only Surviving: 375547 (1.75%) Dropped: 1245929 (5.81%)
TrimmomaticPE: Completed successfully
记住指定接头文件一定要用全路径哦!!!
可以看到它使用了自带的文件TruSeq3-PE.fa里面的接头 TACACTCTTTCCCTACACGACGCTCTTCCGATCT其实只是 TruSeq Universal Adapter (可以在https://github.com/csf-ngs/fastqc/blob/master/Contaminants/contaminant_list.txt 找到接头信息)的后半段,直接在R1测序文件里面搜索可以看到,距离AAAAAAAAAAAAATTTTTTTTTTTTTTTTT这样的字符串和它的 接头 TACACTCTTTCCCTACACGACGCTCTTCCGATCT之间还有序列:

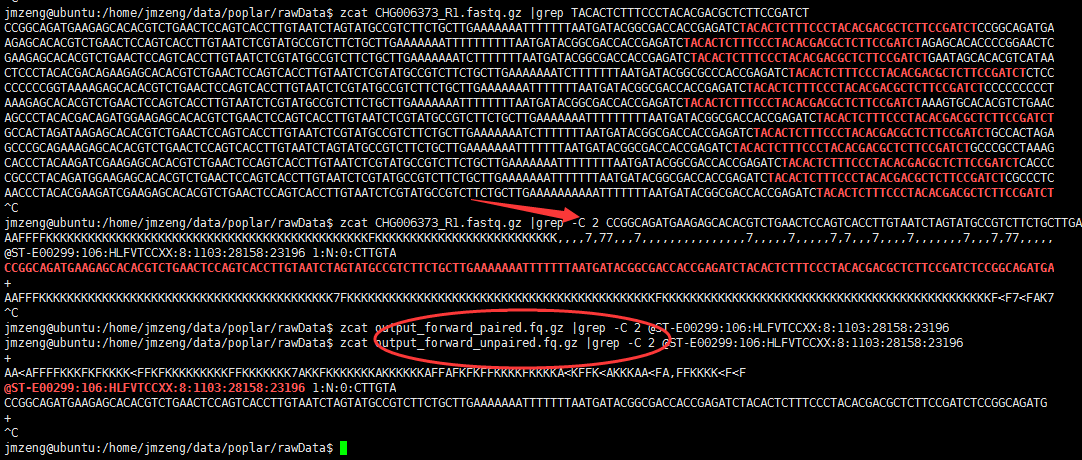
比如我们拿第一个序列举例,可以看到第一条序列被trimmomatic丢到了output_forward_unpaired.fq.gz,它就懒得给它去除接头了,因为右端序列更可怜!
检查文件,发现有的地方是根据质量值来去除的,因为跟接头没有半毛钱关系!

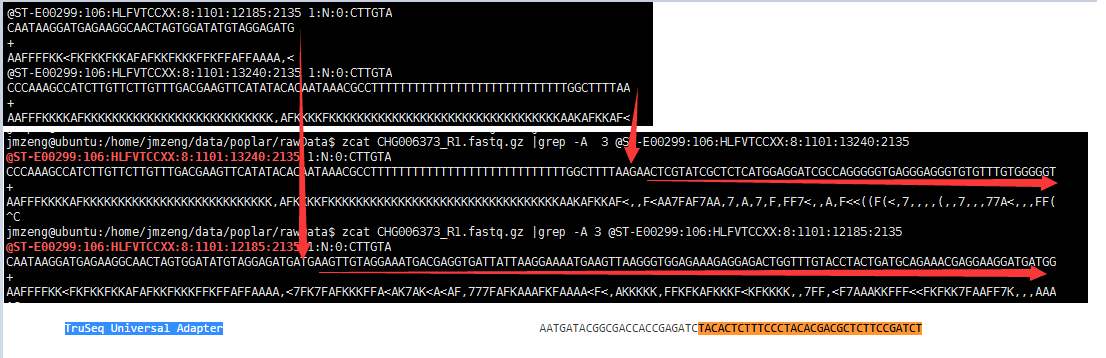
因为它是接头和低质量碱基一起去除,我很难探究它到底是如何去除接头的,非常郁闷,但是它对illumina的数据效果非常好!因为去除的百分比很高。