我以前写过bedtools和htseq-counts的教程,它们都可以用来对比对好的bam文件进行计数,正好群里有小伙伴问我它们的区别,我就简单做了一个比较,大家可以先看看我以前写的软件教程。写的有的挫:
言归正传,我这里没精力去探究它们的具体原理,只是看看它们数一个read是否属于某个基因的时候,区别在哪里,大家看下图:
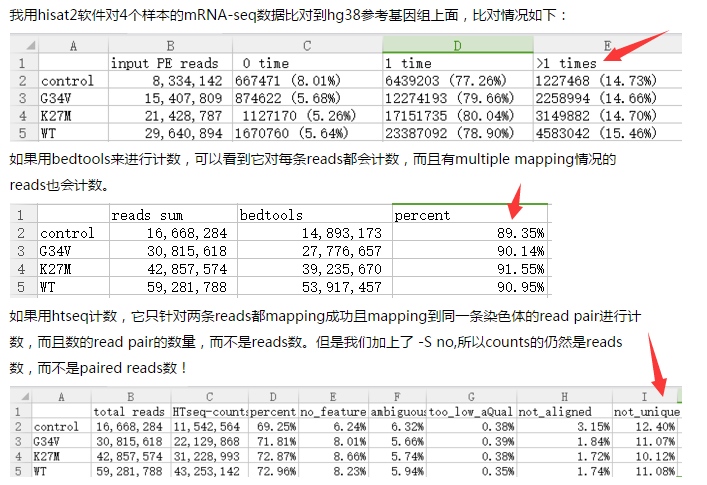
很明显,bedtools不管三七二十一,只要你的reads比对到基因组的坐标跟目的基因坐标有交叉,就算你一个reads,不需要管你是不是multiple mapping的。
但是htseq就谨慎很多,而且还可以挑选model,一般来说,它会把multiple mapping的reads归类到 not unique aligned里面。
而且,大家做完分析,一定要再三检查,很明显人家hisat告诉你的mapping rate高达90%以上,即使除去那15%左右的multiple mapping,你counts表达量的时候,至少也可以counts 百分之五六十吧!!!
如果出现大数量级的no_feature,你自己就应该明白有问题了!
最后htseq-counts使用的时候有一些参数尤其需要注意:
参考gtf文件可以是gencode或者是ensembl数据库的,但是尤其要注释chr的问题,而且版本问题,gtf/gff格式无所谓。比对后的文件一定要进行sort,推荐一定要sort -n,根据reads的name来sort
-f sam/bam 这个一定要搞清楚,如果对bam文件进行counts,必须保证你服务器的python安装了正确的pysam模块
-r name/pos, 一般情况下我们的bam都是按照参考基因组的pos来sort的,但是这个软件默认却是reads的name,很坑,一般建议重新把bam文件sort一下,而不是选择 -r pos,因为-r pos实在是太消耗内存了。
-s yes/no/reverse, 这也是巨坑的参数,默认是yes,一般人拿到的数据都是no,所以千万要注意!!!
-t 选择gff/gtf文件的第3列,一般是exon,也可以是gene,transcript ,这个很少调整的。
-i 这个需要修改,不然默认是ensembl的基因ID,一般人看不懂,可以改为gene_name,前提是你的gff文件里面有gene_name这个属性。
其余的就不需要修改了。
我的代码如下:
nohup samtools view control.Nsort.bam | ~/.local/bin/htseq-count -f sam -s no -i gene_name - ~/reference/gtf/gencode/gencode.v25lift37.annotation.gtf 1>control.geneCounts 2>control.HTseq.log &nohup samtools view G34V.Nsort.bam | ~/.local/bin/htseq-count -f sam -s no -i gene_name - ~/reference/gtf/gencode/gencode.v25lift37.annotation.gtf 1>G34V.geneCounts 2>G34V.HTseq.log &nohup samtools view K27M.Nsort.bam | ~/.local/bin/htseq-count -f sam -s no -i gene_name - ~/reference/gtf/gencode/gencode.v25lift37.annotation.gtf 1>K27M.geneCounts 2>K27M.HTseq.log &nohup samtools view WT.Nsort.bam | ~/.local/bin/htseq-count -f sam -s no -i gene_name - ~/reference/gtf/gencode/gencode.v25lift37.annotation.gtf 1>WT.geneCounts 2>WT.HTseq.log &