熟悉我的人都知道RNA-seq是我的拿手好戏啦!
但是,今天处理了一个公共数据,比对率低的惊人!
是测序数据质量不好?
难道grcm38与mm10有差别?
还是比对工具的默认参数不行?
请看下去,看看老司机是如何翻车的!
数据比较新,是理所当然的认为测序数据肯定是OK的:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE81916
下载sra数据,转换为fastq我就不讲解了!
Written 30468155 spots for SRR3589959.sra
Written 52972617 spots for SRR3589960.sra
Written 36763726 spots for SRR3589961.sra
Written 43802631 spots for SRR3589962.sra
我用的是hisat2工具来比对,一般情况下我就用默认参数啦!
reference=/home/jianmingzeng/reference/index/hisat/grcm38/genome
~/biosoft/HISAT/current/hisat2 -p 5 -x $reference -U SRR3589959.fastq -S control_1.sam 2>control_1.log
~/biosoft/HISAT/current/hisat2 -p 5 -x $reference -U SRR3589960.fastq -S control_2.sam 2>control_2.log
~/biosoft/HISAT/current/hisat2 -p 5 -x $reference -U SRR3589961.fastq -S Akap95_1.sam 2>Akap95_1.log
~/biosoft/HISAT/current/hisat2 -p 5 -x $reference -U SRR3589962.fastq -S Akap95_2.sam 2>Akap95_2.log
ls *sam |while read id;do (nohup samtools sort -n -@ 5 -o ${id%%.*}.Nsort.bam $id &);done
但是让我意外的是比对率出奇的低~~~
0.48% overall alignment rate
0.62% overall alignment rate
0.48% overall alignment rate
0.49% overall alignment rate
起初我怀疑是参考基因组用错了,但是我查看了GEO里面的介绍,的确是mouse的ESC,所以我用grcm38没有问题呀!
然后我怀疑是测序数据质量的问题,但是质量再差也不会导致如此低的比对率呀~~~
所以我还是用fastqc检查了一下:
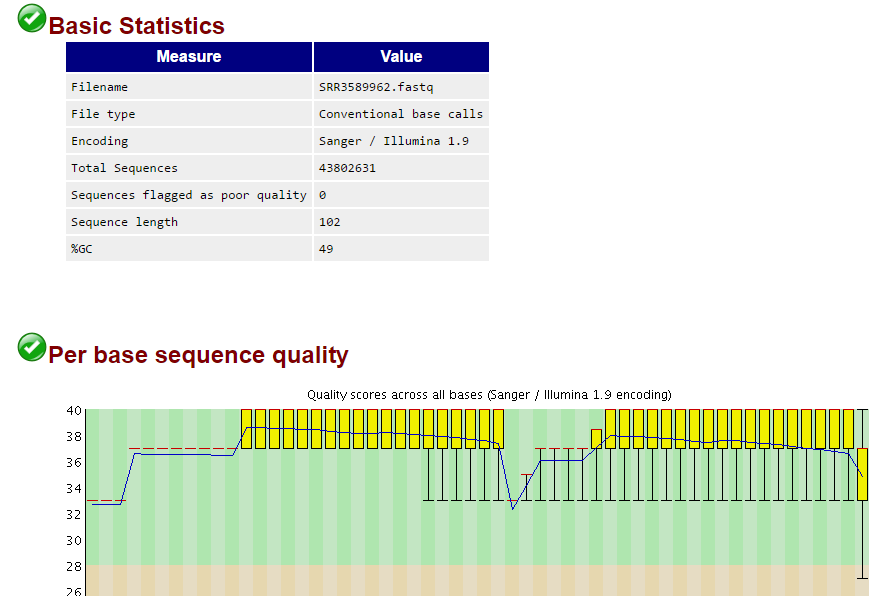
果然,质量值好到爆!!!!
而且我抽取了几条序列去blat一下,发现也可以比对呀,而且很明显是跨越intron的比对,超级经典的RNA-seq数据呀!!!
( 其实我这个blat结果也没有看仔细,正常的reads不应该被截成比对到基因组的正负链的,这其实预示着我把PE序列拼接了。)
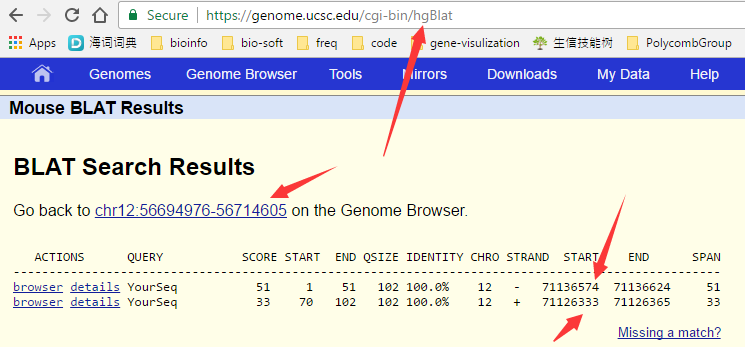
那么就是我hisat2这个步骤的问题咯,我首先怀疑是不是我下载hisat的index搞错了,虽然看起来我命名是grcm38,但是有可能是我下载错误!我打开了sam文件看了看开头:
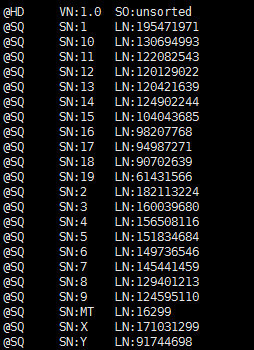
貌似的确是mouse基因组的染色体长度呀!很诡异,而且我清楚的记得,我下载的就是mouse的基因的索引呀!
难道grcm38与mm10有差别?
我就先用bowtie2测试一下mm10吧,毕竟我还没有hisat2的mm10的index呀!
head -1000 SRR3589959.fastq >tmp.fq
~/biosoft/bowtie/bowtie2-2.2.9/bowtie2 -p 6 -x ~/reference/index/bowtie/mm10 -U tmp.fq -S tmp.sam
结果我挑出来的这1000条序列,全军覆没了,0.00% overall alignment rate,我傻眼了!
没办法呀,逼着我换hg19参考基因组看看:
~/biosoft/bowtie/bowtie2-2.2.9/bowtie2 -p 6 -x ~/reference/index/bowtie/hg19 -U tmp.fq -S tmp.hg19.sam
仍然是全军覆没了,0.00% overall alignment rate,继续傻眼!
回过头看了看fastqc的报告,发现前面10个碱基的确有问题的!如果只是对RNA-seq进行定量,可能需要trim掉,但是,我以前从来不trim,照样不影响比对呀
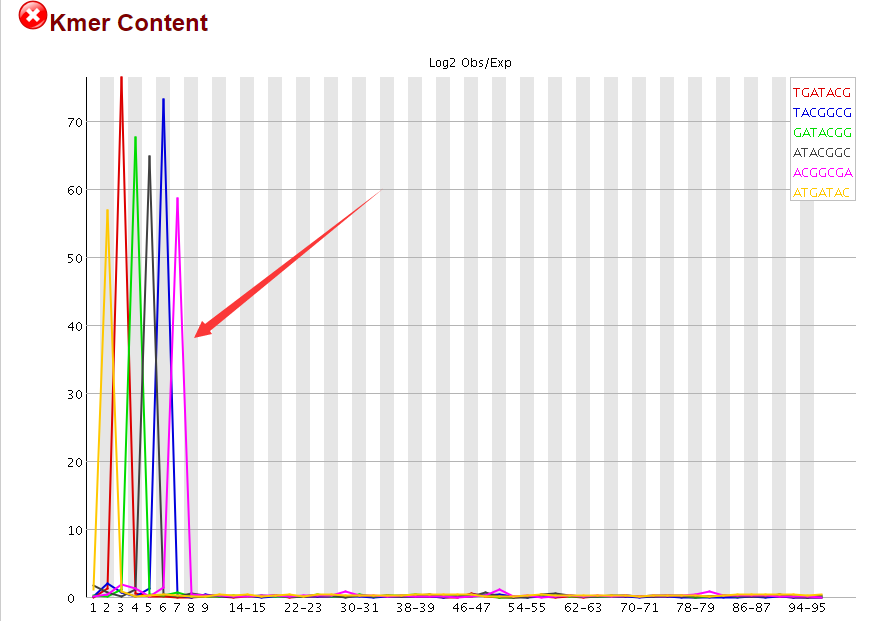
不过,暂时看到这个问题,我就试着解决一下吧,先从这个思路来,
而且比对工具里面本来就有这个选项,没必要自己来trim的!具体参数见:https://ccb.jhu.edu/software/hisat2/manual.shtml
-5/--trim5 <int> trim <int> bases from 5'/left end of reads (0)
-3/--trim3 <int> trim <int> bases from 3'/right end of reads (0)
所以我加上了-p 6 -5 10 -3 10 --local 参数,比对人,可以拿到35.60% overall alignment rate,比对mouse,可以拿到98.80% overall alignment rate ,我勒个去,问题出来了,看起来好像是应该trim掉呀。以前的万能默认参数不行了!!!!
但是有个问题,虽然我用local模式都比对上了,但是首先100bp的reads我切成了80,而且都是40M,40S,说明只有reads的一般成功比对到了参考基因组序列呀!!!!
我然后用同样的参数,我测试了hisat2工具,但是hisat2里面压根就没有local的选项,仅仅是trim一下,对比对的改善毫无意义,所以重点在于--local这个参数,但它只是表象,本质还是这个测序数据出问题了!
数据为什么会出问题呢?
我再回过头看了看测序数据的fastqc报告,我勒个去,这么重要的图我居然忽略掉了,再联想到前面的40M,40S我瞬间明白了,这肯定是一个双端测序,被我搞成 了单端测序数据!
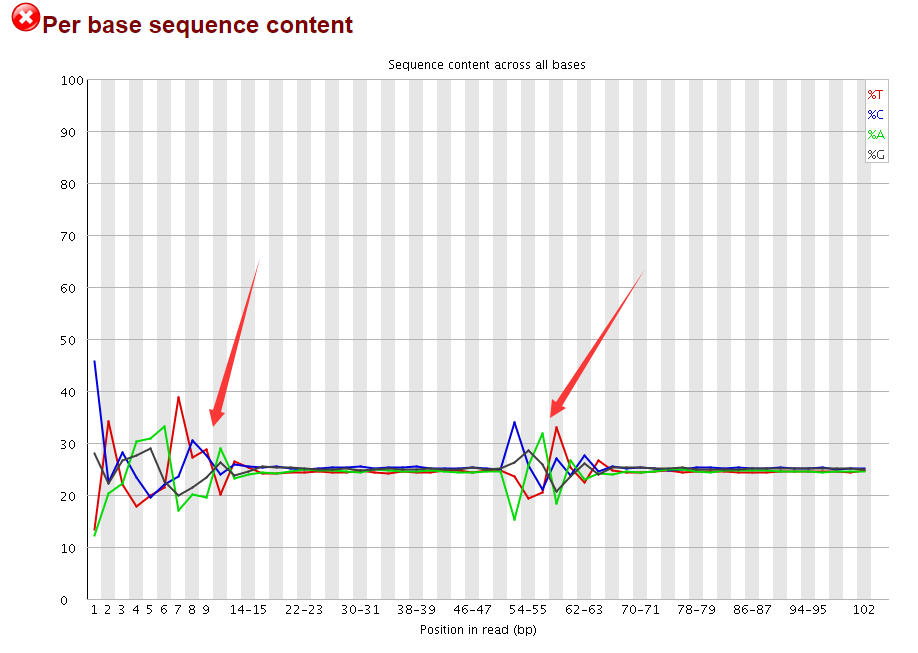
而且我再去GEO介绍上面看,上面赫然写着PAIRED!!!!我死也想不明白,我明明是加了--split-3 参数呀,为什么sra转换成fastq会出这么明显的错误呢?
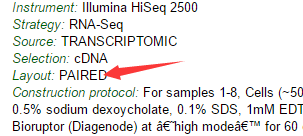
然后我检查我的脚本,马勒戈壁,我自己从我博客里面复制了我的代码,

唯一值得你看的就是这个图
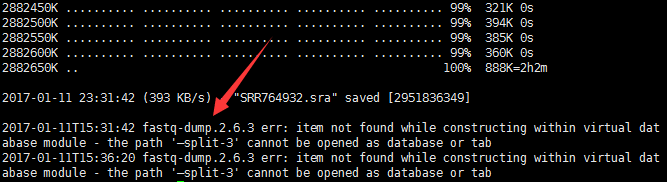
是-- 不是— ,全角半角害死人呀,而且这个参数不识别它居然不报错,而是忽略我 参数!!!
是-- 不是— ,全角半角害死人呀,而且这个参数不识别它居然不报错,而是忽略我 参数!!!
是-- 不是— ,全角半角害死人呀,而且这个参数不识别它居然不报错,而是忽略我 参数!!!
更要命的是我把wget跟fastq-dump一起运行的,而wget会给出一大堆的log日志,我都懒得看,结果,把fastq-dump的报错日志给掩盖了。
这就是老司机翻车的全部故事,希望你们引以为戒!
因为前面一直处理的是单端的数据,所以这个错误没有被发现。
我痛恨我博客的脚本了,而且我痛恨--这样的参数设置!
下面是我修改后的代码!!!
cut -f 3 config.txt |while read id ; do wget $id 2>/dev/null ;done
ls *sra |while read id; do ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastq-dump --gzip --split-3 $id;done
老司机现在很伤心,一天的功夫白费了。
因为我已经把sra数据删除了,想重来一次的机会都不给我~~~
又要重新下载一次,好惨啊!!!!
总结一下吧:
QC这一步骤非常重要,不能太马虎!
原始数据不要随意删除,给自己一次重新来过的机会。