请不要直接拷贝我的代码,需要自己理解,然后打出来,思考我为什么这样写代码。
软件请用最新版,尤其是samtools等被我存储在系统环境变量的,考虑到读者众多,一般的软件我都会自带版本信息的!
我用两个小时,不代表你是两个小时就学会,有些朋友反映学了两个星期才 学会,这很正常,没毛病,不要异想天开两个小时就达到我的水平。
MeDIP-seq 跟ChIP-seq的分析手段是一模一样的,同理hMeDIP-seq,caMeDIP-seq等等,都没有本质上的区别,只是用的抗体不一样而已,请自行搜索基础知识,我只讲数据分析。
一个RNA-seq实战-超级简单-2小时搞定!
请先看看我前面写的系列,对我而言很简单,因为软件我都安装了,数据我都下载好了,代码我都看得懂,对你,不一定简单,有朋友反映学了两个星期才弄懂,但至少,是可以弄懂的!
paper是Dnmt3L antagonizes DNA methylation at bivalent promoters and favors DNA methylation at gene bodies in ESCs.:https://www.ncbi.nlm.nih.gov/pubmed/24074865 发表在2013年CELL杂志上面,值得重复!
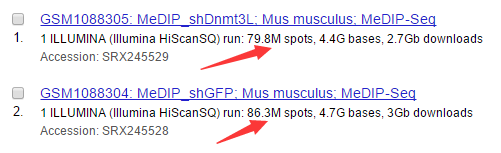
首先下载raw data数据:
wget ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/sra/SRP/SRP018/SRP018845/SRR764931/SRR764931.sra
wget ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/sra/SRP/SRP018/SRP018845/SRR764932/SRR764932.sra
ls *sra |while read id; do ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastq-dump --split-3 $id;done
用fastqc看了看数据质量,发现质量非常赞,我就不需要过滤reads了。代码如下:
ls *fastq |xargs ~/biosoft/fastqc/FastQC/fastqc -t 10
如果要过滤,就用下面的代码:
ls *.fastq | while read id
do
~/biosoft/sickle/sickle-master/sickle se -t sanger -g -f $id -o ${id%%.*}.trimmed.fq.gz
done
首先用bowtie2软件把测序得到的fastq文件比对到mm10参考基因组上面,就两个数据,我就不写循环了!
对于这种没有control的数据,我们可以直接把peaks-calling 4部曲一起搞定的!
对比对好的bam文件, 就可以直接用MACS软件来找peaks啦:
首先对这些bam文件批量转换成bw文件。然后批量画图
~/biosoft/bowtie/bowtie2-2.2.9/bowtie2 -p 6 -x ~/reference/index/bowtie/mm10 -U SRR764931.fastq | samtools sort -O bam -o shDnmt3L.bam
## 比对率很高,分别是96.67%(shDnmt3L) 和96.59%(shGFP),这比对率没得说了,非常赞!
samtools index shDnmt3L.bam
~/.local/bin/macs2 callpeak -t shDnmt3L.bam -m 10 30 -p 1e-5 -f BAM -g mm -n shDnmt3L 2>shDnmt3L.masc2.log
bamCoverage -b shDnmt3L.bam -o shDnmt3L.bw ## 这里有个参数,-p 10 --normalizeUsingRPKM
computeMatrix reference-point --referencePoint TSS -b 10000 -a 10000 -R ~/annotation/CHIPseq/mm10/ucsc.refseq.bed -S shDnmt3L.bw --skipZeros -o matrix1_shDnmt3L_TSS.gz
plotHeatmap -m matrix1_shDnmt3L_TSS.gz -out shDnmt3L.png
就两个数据,我就没有写循环了,现在你肯定能看懂了吧!
分析,就这样介绍咯!