可变剪切是指mRNA前体以多种方式将exon连接在一起的过程。 由于可变剪切使一个基因产生多个mRNA转录本,不同mRNA可能翻译成不同蛋白。
可变剪切背景知识
转录组一般是指从细胞或组织的基因组所转录出来的RNA的总和,包括编码蛋白质的mRNA和各种非编码RNA(rRNA,tRNA,snRNA,snoRNA,lncRNA,microRNA等)。真核生物的基因结构是不连续的,如下图:
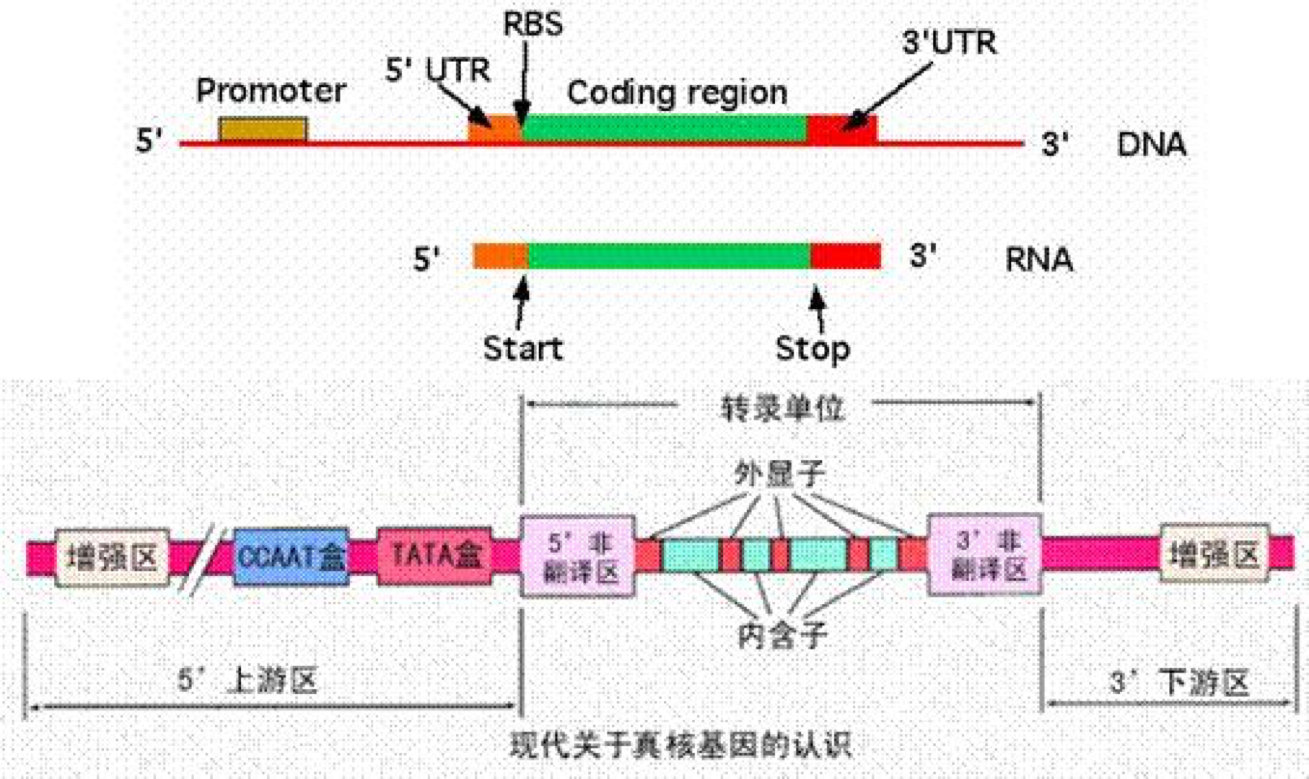
其基因组最初的转录产物其实并不是成熟的mRNA分子,而是它的前体pre-mRNA,那么怎么变成成熟的mRNA呢,就需要从pre-mRNA中将非编码蛋白质的内含子(intron)切除,然后拼接剩下的编码蛋白质的外显子(exon)。但实际上,在这个过程中,有多种多样的前切和拼接方式,从而产生不同的剪切异构体,也就咱们要说的可变剪切。
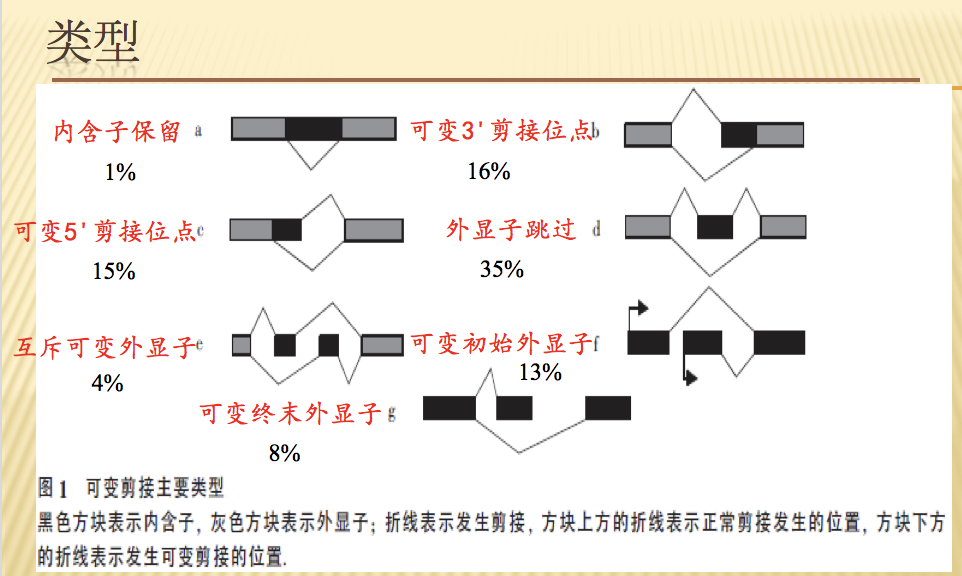
可变剪切的形式复杂多样,大致可以分为5大类。
- 第一类是外显子跳跃型(exon skipping),发生跳跃的外显子和其两侧的内含子都被剪切掉,上游和下游的外显子被直接连着一起保留在剪切后的产物中。
- 第二类是内含子滞留型(intron retention),某一段核苷酸序列在一个剪切体中是外显子的一部分,而在与之对照的剪切体中却是内含子而被剪切掉。
- 第三类是可变5’或3’端剪切(alternative 5’ss splice or alternative 3’ss splice,其中5’ss称供体位点,3’ss称受体位点),和与它对照的另一个剪切体相比,发生剪切的位点在5’或3’端不同,除此,其他剪切选择一致。
- 第四类是转录起始区域可变剪切(alternative TSS),发生剪切的位点在转录起始区域,即与之对应的另一个剪切体除转录起始位点不同外,其余一致。
- 第五类是转录终止区域可变剪切(alternative TTS),与第四类对应,发生剪切的位点只是在转录终止位点不同。
软件算法
比较旧的分析可变剪切的软件主要有SpliceR、SpliceGrapher、ASprofile以及Splicing Express等,它们是基于cufflinks软件的结果,将reads回帖到基因组序列后,根据位置和长度及结构信息,来确定或预测可能的剪切体的类型。目前主流已经不再使用tophat+cufflinks流程了。
SGSeq流程
这里介绍一下SGSeq软件,输入文件是bam,但是需要用支持转录组数据比对的工具得到的bam文件,比如
- GSNAP (T. D. Wu and Nacu 2010)
- HISAT (Kim, Langmead, and Salzberg 2015)
- STAR (Dobin et al. 2013)
其实是需要bam文件里面有XS 这样的标记!
SGSeq包的安装说明,使用方法都可以见官网:
| HTML | R Script | SGSeq |
|---|---|---|
| Reference Manual | ||
| Text | NEWS |
需要bam文件
安装好包之后可以看到附带的数据,如下:
jianmingzengs-iMac:IGV_2.3.98 jmzeng$ cd /Library/Frameworks/R.framework/Versions/3.4/Resources/library/SGSeq/extdata/bams/
jianmingzengs-iMac:bams jmzeng$ ls -lh
total 1952
-rw-r--r-- 1 jmzeng admin 54K Nov 1 01:26 N1.bam
-rw-r--r-- 1 jmzeng admin 43K Nov 1 01:26 N1.bam.bai
-rw-r--r-- 1 jmzeng admin 86K Nov 1 01:26 N2.bam
-rw-r--r-- 1 jmzeng admin 43K Nov 1 01:26 N2.bam.bai
-rw-r--r-- 1 jmzeng admin 75K Nov 1 01:26 N3.bam
-rw-r--r-- 1 jmzeng admin 43K Nov 1 01:26 N3.bam.bai
-rw-r--r-- 1 jmzeng admin 92K Nov 1 01:26 N4.bam
-rw-r--r-- 1 jmzeng admin 43K Nov 1 01:26 N4.bam.bai
-rw-r--r-- 1 jmzeng admin 75K Nov 1 01:26 T1.bam
-rw-r--r-- 1 jmzeng admin 43K Nov 1 01:26 T1.bam.bai
-rw-r--r-- 1 jmzeng admin 90K Nov 1 01:26 T2.bam
-rw-r--r-- 1 jmzeng admin 43K Nov 1 01:26 T2.bam.bai
-rw-r--r-- 1 jmzeng admin 65K Nov 1 01:26 T3.bam
-rw-r--r-- 1 jmzeng admin 43K Nov 1 01:26 T3.bam.bai
-rw-r--r-- 1 jmzeng admin 75K Nov 1 01:26 T4.bam
-rw-r--r-- 1 jmzeng admin 43K Nov 1 01:26 T4.bam.bai
这些bam文件之所以这么小,就是因为作者只是截取了hg19的部分数据,坐标是16 [87362942, 87425708]
需要注释文件
需根据bioconductor里面的txdb对象来构建比对文件的参考基因组,参考注释信息。如果是hg19的可以如下:
library(TxDb.Hsapiens.UCSC.hg19.knownGene)
txdb <- TxDb.Hsapiens.UCSC.hg19.knownGene
txdb <- keepSeqlevels(txdb, "chr16")
seqlevelsStyle(txdb) <- "NCBI"
txf_ucsc <- convertToTxFeatures(txdb)
txf_ucsc <- txf_ucsc[txf_ucsc %over% gr]
head(txf_ucsc)
type(txf_ucsc)
head(txName(txf_ucsc))
head(geneName(txf_ucsc))
主要就是通过convertToTxFeatures()函数把 GRanges 对象转化成了一个TxFeatures对象,用来标记下面5种类型:
- J (splice junction)
- I (internal exon)
- F (first/5′′-terminal exon)
- L (last/5′′-terminal exon)
- U (unspliced transcript).
再用 convertToSGFeatures() 函数把TxFeatures对象转化成SGFeatures 对象,用来标记
- J (splice junction)
- E (disjoint exon bin)
- D (splice donor site)
- A (splice acceptor site).
运行SGSeq软件
sgfc_ucsc <- analyzeFeatures(si, features = txf_ucsc)
sgfc_ucsc
因为软件包自带的数据非常小,所以很容易就运行完毕,不知道真实情况下我的16G的bam文件会处理多久。
探索处理结果
也是全部在R语言里面运行即可,下面的这些函数用来探索分析结果,这些表达矩阵就写明了每个基因的每个外显子的表达量以及两个外显子中间夹着的内含子的表达情况。
也就是说该软件在R里面就对所有的genomic features 进行了reads的计数。
colData(sgfc_ucsc)
rowRanges(sgfc_ucsc)
head(counts(sgfc_ucsc))
head(FPKM(sgfc_ucsc))
可变剪切形式的可视化
挑选其中一个基因,可视化表达差异情况
df <- plotFeatures(sgfc_ucsc, geneID = 1)
# 下面是复杂一点的可视化
sgfc_pred <- analyzeFeatures(si, which = gr)
head(rowRanges(sgfc_pred))
sgfc_pred <- annotate(sgfc_pred, txf_ucsc)
head(rowRanges(sgfc_pred))
df <- plotFeatures(sgfc_pred, geneID = 1, color_novel = "red")
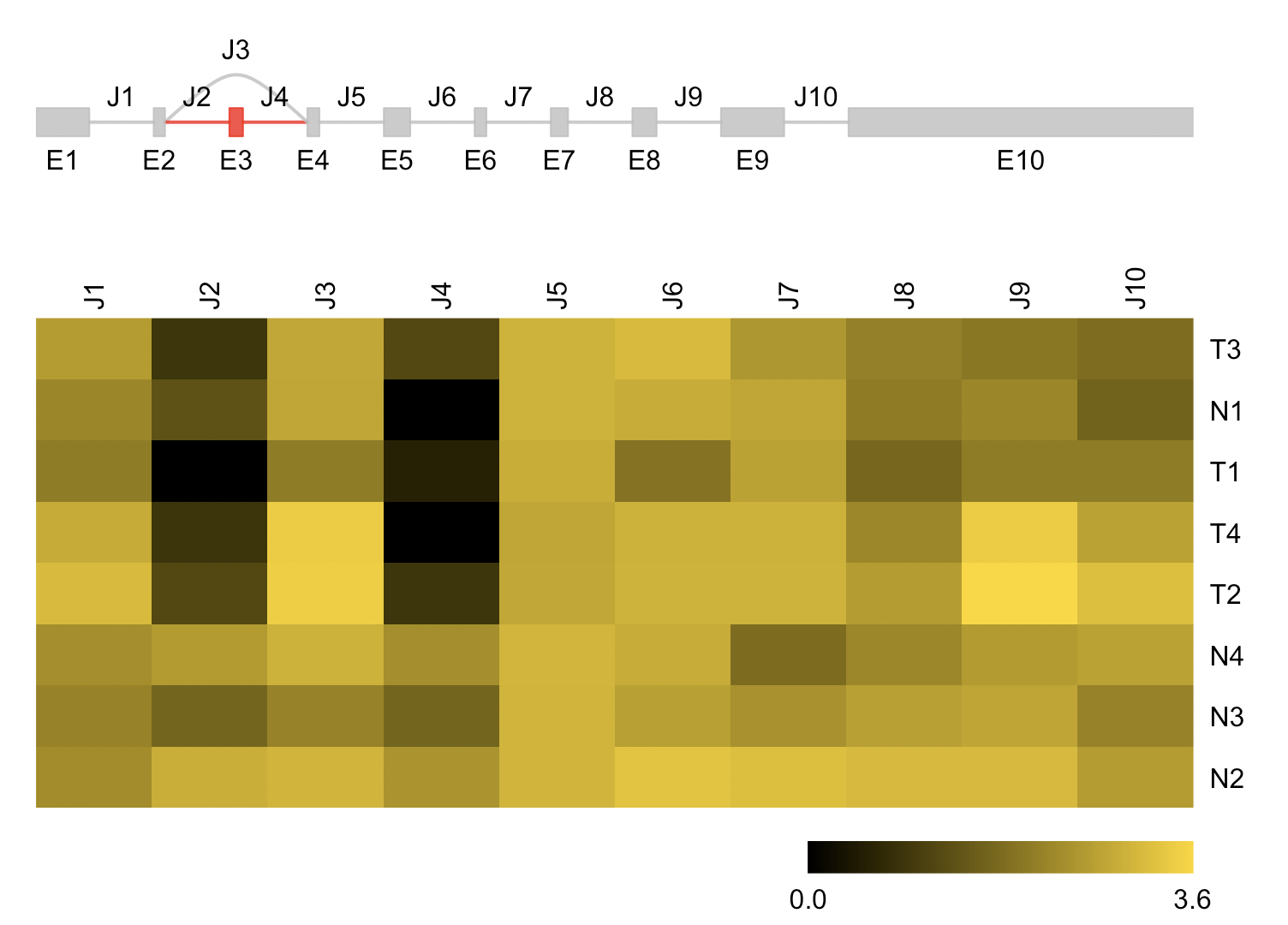 这个是作者精选挑选的特殊的例子用来展现软件的成功,事实上应该是先全局检查哪些可变剪切存在,然后输出
这个是作者精选挑选的特殊的例子用来展现软件的成功,事实上应该是先全局检查哪些可变剪切存在,然后输出
## 下面是另外一个展现模式:
par(mfrow = c(5, 1), mar = c(1, 3, 1, 1))
plotSpliceGraph(rowRanges(sgfc_pred), geneID = 1, toscale = "none", color_novel = "red")
for (j in 1:4) {
plotCoverage(sgfc_pred[, j], geneID = 1, toscale = "none")
}
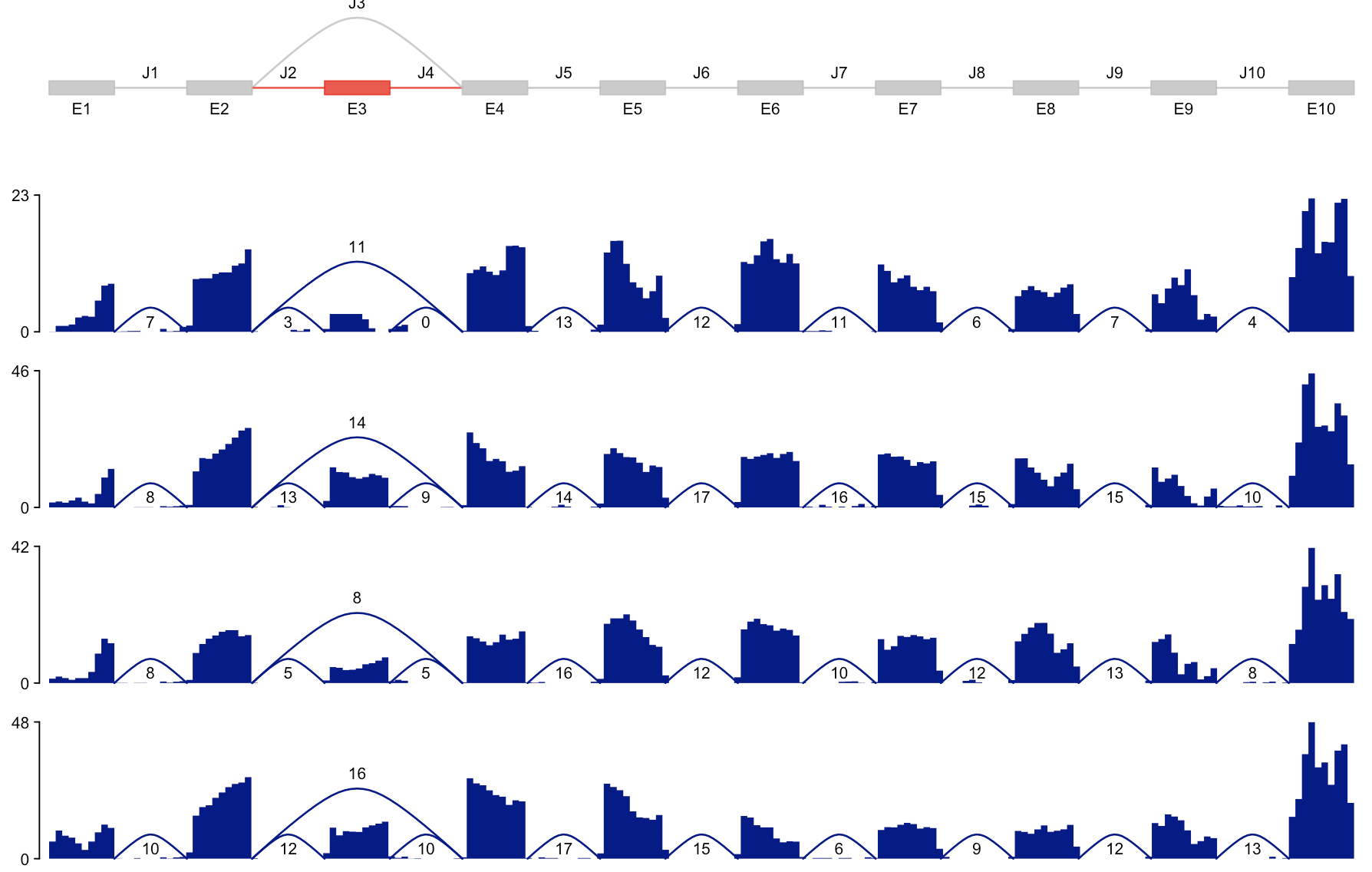
从可变剪切预测结果里面鉴别剪切体
Instead of considering the full splice graph of a gene, the analysis can be focused on individual splice events. Function analyzeVariants() recursively identifies splice events from the graph, obtains representative counts for each splice variant, and computes estimates of relative splice variant usage, also referred to as ‘percentage spliced in’ (PSI or Ψ) (Venables et al. 2008, Katz et al. (2010)). (涉及到了一个算法的问题)
sgvc_pred <- analyzeVariants(sgfc_pred)
sgvc_pred
mcols(sgvc_pred)
variantFreq(sgvc_pred)
plotVariants(sgvc_pred, eventID = 1, color_novel = "red")
library(BSgenome.Hsapiens.UCSC.hg19)
seqlevelsStyle(Hsapiens) <- "NCBI"
vep <- predictVariantEffects(sgv_pred, txdb, Hsapiens)
vep