linux都无法掌握,就 不要学生信了,没有意义,需要时间和努力才能掌握linux,也不要妄想就接触了linux一天就做完下面的题目,需要购买linux书籍加上一个月的持续学习!
重点:
(去可视化概念+练习) 了解 命令+参数+文件 的模式
基础知识:cd -, cd .. , cd -, history, !5 , /home/ , /tmp/ , >,&,jobs,nohup 1,2,0 文件目录操作:ls,cd,pwd,mkdir,rm,mv,cp,touch,head,tail,less,more 系统管理: df,du,top,free,ps,ipconfig,netstat,ssh,scp, 用户权限:chown,chgrp,groups,ls
文本操作:awk,grep,sed,paste,cat,diff,wc,vi
使用腾讯云实验室的linux服务器:https://cloud.tencent.com/developer/labs/lab/10000
参考 生物信息学常见1000个软件的安装代码! 来安装软件。
如果学完了,理论上你看下面的总结应该是有茅塞顿开的感觉。
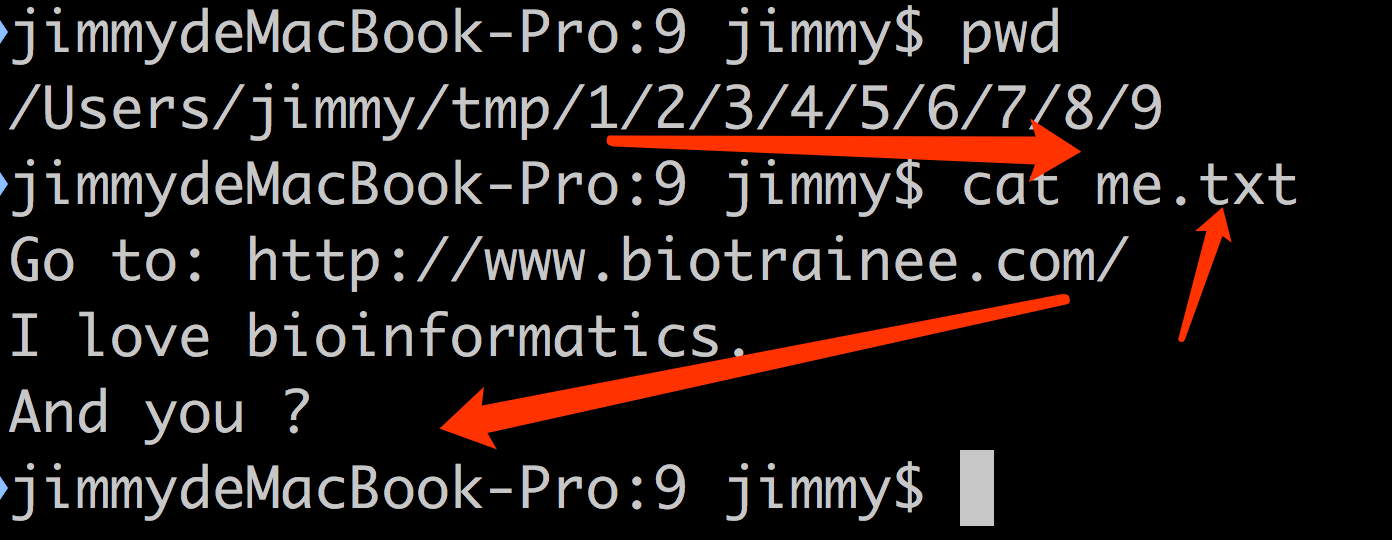
一、在任意文件夹下面创建形如 1/2/3/4/5/6/7/8/9 格式的文件夹系列。
二、在创建好的文件夹下面,比如我的是 /Users/jimmy/tmp/1/2/3/4/5/6/7/8/9 ,里面创建文本文件 me.txt
三、在文本文件 me.txt 里面输入内容:
Go to: http://www.biotrainee.com/
I love bioinfomatics.
And you ?
前三题效果如下:
 、删除上面创建的文件夹
、删除上面创建的文件夹 1/2/3/4/5/6/7/8/9 及文本文件 me.txt
五、在任意文件夹下面创建 folder1~5这5个文件夹,然后每个文件夹下面继续创建 folder1~5这5个文件夹,效果如下:
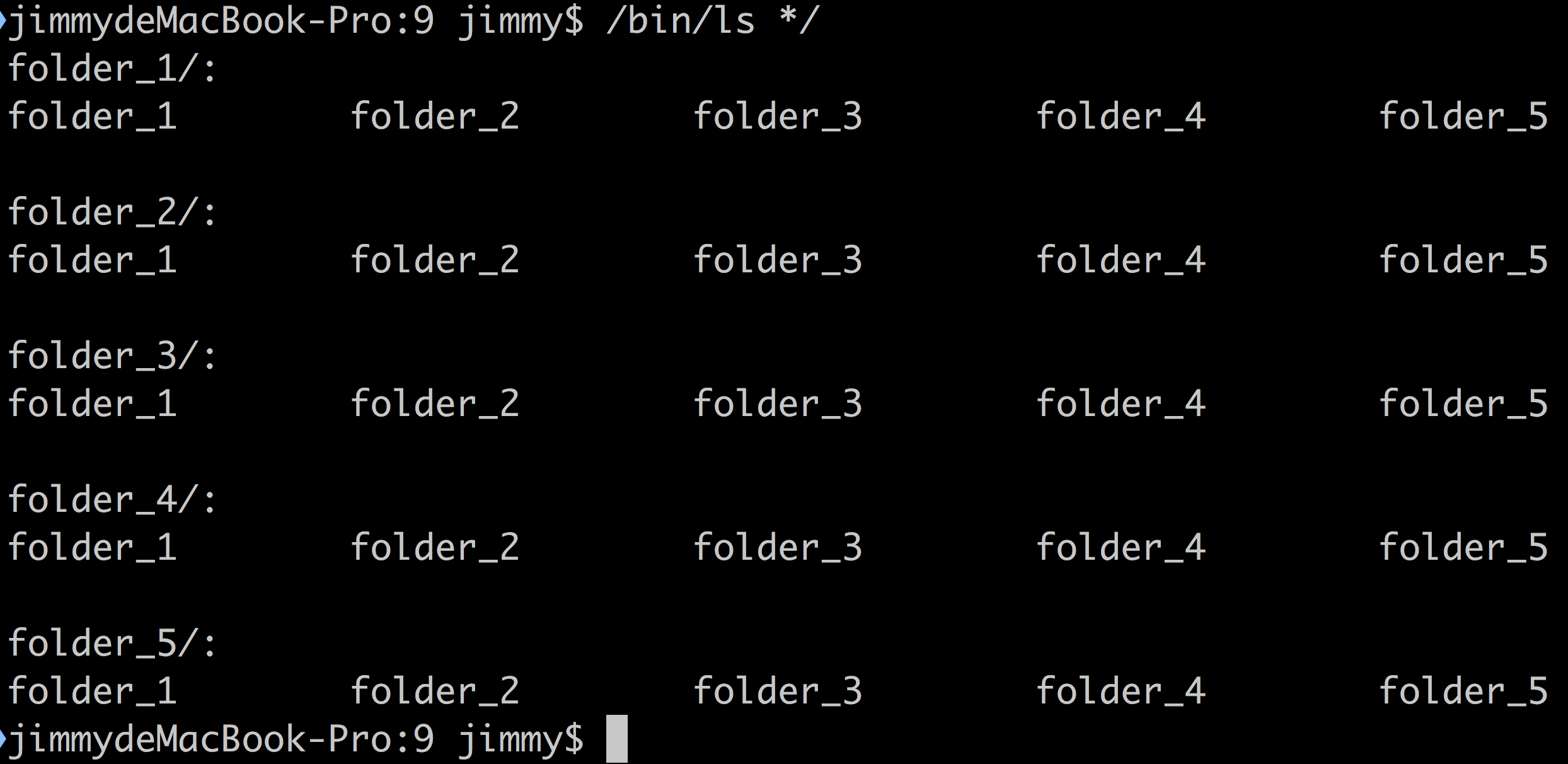
六、在第五题创建的每一个文件夹下面都 创建第二题文本文件 me.txt ,内容也要一样。(这个题目难度超纲,建议一个月后再回过头来做)
七,再次删除掉前面几个步骤建立的文件夹及文件
八、下载 http://www.biotrainee.com/jmzeng/igv/test.bed 文件,后在里面选择含有 H3K4me3 的那一行是第几行,该文件总共有几行。
九、下载 http://www.biotrainee.com/jmzeng/rmDuplicate.zip 文件,并且解压,查看里面的文件夹结构
十、打开第九题解压的文件,进入 rmDuplicate/samtools/single 文件夹里面,查看后缀为 .sam 的文件,搞清楚 生物信息学里面的SAM/BAM 定义是什么。
十一、安装 samtools 软件
十二、打开 后缀为BAM 的文件,找到产生该文件的命令。 提示一下命令是:
/home/jianmingzeng/biosoft/bowtie/bowtie2-2.2.9/bowtie2-align-s --wrapper basic-0 -p 20 -x /home/jianmingzeng/reference/index/bowtie/hg38 -S /home/jianmingzeng/data/public/allMouse/alignment/WT_rep2_Input.sam -U /tmp/41440.unp
十三题、根据上面的命令,找到我使用的参考基因组 /home/jianmingzeng/reference/index/bowtie/hg38 具体有多少条染色体。
十四题、上面的后缀为BAM 的文件的第二列,只有 0 和 16 两个数字,用 cut/sort/uniq等命令统计它们的个数。
十五题、重新打开 rmDuplicate/samtools/paired 文件夹下面的后缀为BAM 的文件,再次查看第二列,并且统计
十六题、下载 http://www.biotrainee.com/jmzeng/sickle/sickle-results.zip 文件,并且解压,查看里面的文件夹结构, 这个文件有2.3M,注意留心下载时间及下载速度。
十七题、解压 sickle-results/single_tmp_fastqc.zip 文件,并且进入解压后的文件夹,找到 fastqc_data.txt 文件,并且搜索该文本文件以 >>开头的有多少行?
十八题、下载 http://www.biotrainee.com/jmzeng/tmp/hg38.tss 文件,去NCBI找到TP53/BRCA1等自己感兴趣的基因对应的 refseq数据库 ID,然后找到它们的hg38.tss 文件的哪一行。
https://www.ncbi.nlm.nih.gov/gene/7157
十九题、解析hg38.tss 文件,统计每条染色体的基因个数。
二十题、解析hg38.tss 文件,统计NM和NR开头的熟练,了解NM和NR开头的含义。