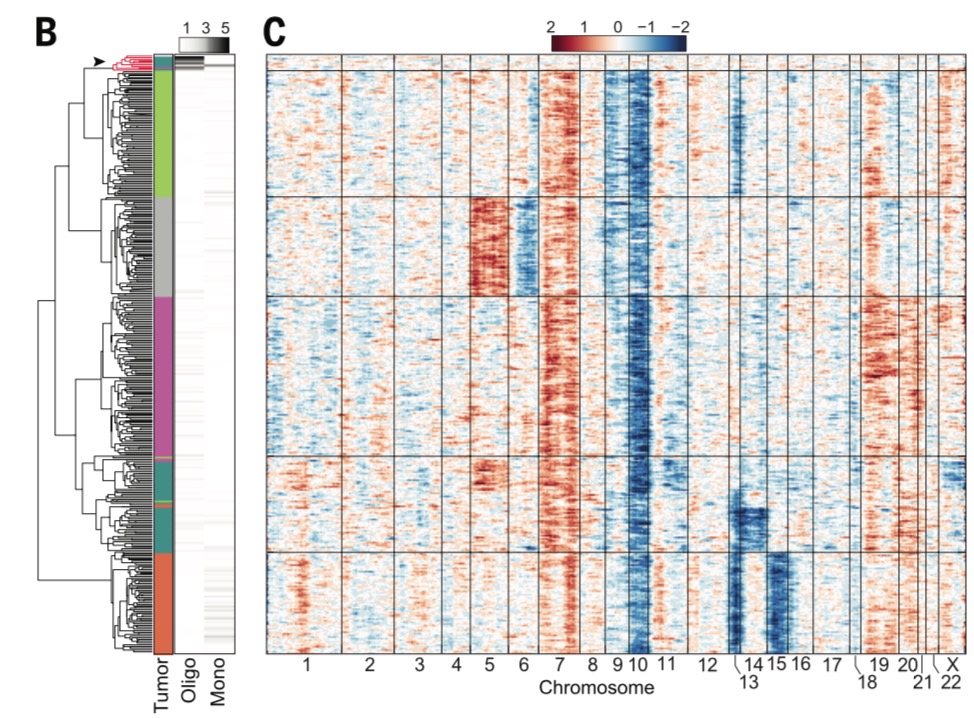
前面我们提到过broad开发了工具来对单细胞转录组数据计算CNV及绘制热图,因为这个方法学本来就是他们建立起来的。
- 单细胞转录组数据分析CNV
- 使用broad出品的inferCNV来对单细胞转录组数据推断CNV信息
- 使用inferCNV来推断CCLE转录组数据的拷贝数变异
- 使用inferCNV来推断2014的science关于GBM文章的单细胞转录组数据的拷贝数情况
我也拿那个软件在普通的bulk转录组数据,CCLE数据库数据,以及两个单细胞数据集测试了,最后在2014的science关于GBM文章的数据里面验证了,可以说已经学会了该软件的使用,但是只会用软件成不了气候,还是得深入理解原理。
数据预处理
得到如下所示的数据,5个病人的400多个单细胞的近6000个基因的表达矩阵,以及这些基因的坐标信息。
> head(res[,1:8])
gene chr start end MGH264_A01 MGH264_A02 MGH264_A03 MGH264_A04
55 NOC2L 1 944204 959309 0.000000 0.0000000 0.000000 8.280510
61 ISG15 1 1001138 1014541 0.000000 6.1750906 0.000000 0.000000
85 CPSF3L 1 1311585 1324691 2.123605 1.3405256 3.354979 0.000000
91 AURKAIP1 1 1373730 1375495 0.000000 4.9487175 0.000000 0.000000
93 CCNL2 1 1385711 1399328 0.000000 5.6449506 0.000000 0.000000
95 MRPL20 1 1401908 1407313 6.980823 0.8206633 3.243285 7.662195
> table(res$chr)
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
603 439 381 231 310 248 332 207 189 165 319 335 92 212 164 195 297 91 327 153
21 22 23 24
56 104 212 7
下面的文件GBM_for_CNV_input.Rdata我已经制作好了,想重复该流程的朋友,可以发邮件找我申请,我的邮箱地址是 jmzeng1314@163.com 也可以自己去 https://portals.broadinstitute.org/single_cell/study/glioblastoma-intra-tumor-heterogeneity#study-summary 下载这个数据集,自己做预处理。
只需要运行下面代码即可,代码如下
rm(list = ls())
load(file = 'GBM_for_CNV_input.Rdata')
colnames(pos)=c('gene','chr','start','end')
exprSet[1:4,1:4]
head(pos)
exprSet=exprSet[match(pos$gene,rownames(exprSet)),]
res=cbind(pos,exprSet)
head(res[,1:10])
table(res$chr)
基因的表达量在这里是 log(CPM+1)
分染色体以100个基因步长移动计算CNV
可以说看到那个science文章的补充材料里面的methods部分,写出下面代码,就是水到渠成的事情了。
all_cnv <- lapply(split(res,res$chr), function(x){
# x=split(res,res$chr)[[1]]
anno=x[,1:4]
dat=x[,5:434]
if(nrow(dat)>100){
cnv <- lapply(51:(nrow(dat)-50), function(i){
this_cnv <- unlist( lapply(1:ncol(dat), function(j){
sum(dat[(i-50):(i+50),j])/101
}))
return(this_cnv)
})
cnv=do.call(rbind,cnv)
cnv=cbind(anno[51:(nrow(x)-50),],cnv)
# cnv[1:4,1:8]
}else{
return(NULL)
}
})
all_cnv=do.call(rbind,all_cnv)
head(all_cnv[1:4,1:8])
table(all_cnv$chr)
所以每个染色体上面头尾50个基因是没办法计算CNV的,中间的基因的CNV就是它上下100个基因的表达量的平均值,就是这么简单。
把CNV矩阵归一化并且画热图
这里的归一化是基于样本的,而且可以自由设置截断阈值。
library(pheatmap)
## 按照 列(样本)进行归一化
D=t(scale(all_cnv[,5:ncol(all_cnv)] ))
## 归一化之后进行转置,因为我们的热图列是基因,行是样本。
## 根据阈值进行截断
D[D>2]=2
D[D< -2] = -2
dim(D)
colnames(D)=paste0('genes_',1:ncol(D))
rownames(D)=colnames(exprSet)
require(RColorBrewer)
cols <- colorRampPalette(brewer.pal(10, "RdBu"))(256)
library(stringr)
annotation_row = data.frame(
patients=str_split(rownames(D),'_',simplify = T)[,1]
)
rownames(annotation_row) = rownames(D)
annotation_col = data.frame(
chr= factor(all_cnv$chr,levels = unique(all_cnv$chr))
)
rownames(annotation_col) = colnames(D)
png('GBM_CNV_by_jimmy.png')
pdf('GBM_CNV_by_jimmy.pdf')
pheatmap(D,cluster_rows = T,col=rev(cols),
annotation_col=annotation_col,
annotation_row = annotation_row,
cluster_cols = F,show_rownames=F,show_colnames=F)
dev.off()
我比较喜欢用 pheatmap 绘图,在我的生信菜鸟团博客里面还专门讲解过它,非常方便。
出图如下:
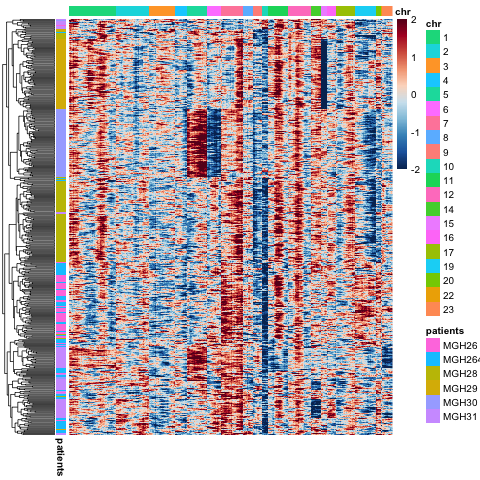
是不是跟那篇paper里面的非常一致呀!