一般来说可以用CV或者MAD来衡量某基因在某些样本的表达变化情况。
标准差与平均数的比值称为变异系数,记为C.V(Coefficient of Variance)。 变异系数又称“标准差率”,是衡量资料中各观测值变异程度的另一个统计量。 当进行两个或多个资料变异程度的比较时,如果度量单位与平均数相同,可以直接利用标准差来比较。
平均绝对误差(Mean Absolute Deviation),又叫平均绝对离差,它是是所有单个观测值与算术平均值的偏差的绝对值的平均。
用下面的代码可以看看,标准差,平均数,变异系数, 平均绝对误差的关系,如下:
library(airway)
library(edgeR)
library(DESeq2)
data(airway)
airway
exprSet=assay(airway)
geneLists=rownames(exprSet)
keepGene=rowSums(cpm(exprSet)>0) >=2
table(keepGene);dim(exprSet)
dim(exprSet[keepGene,])
exprSet=exprSet[keepGene,]
rownames(exprSet)=geneLists[keepGene]
boxplot(exprSet,las=2)
# CPM normalized counts.
exprSet=log2(cpm(exprSet)+1)
boxplot(exprSet,las=2)
mean_per_gene <- apply(exprSet, 1, mean, na.rm = TRUE)
sd_per_gene <- apply(exprSet, 1, sd, na.rm = TRUE)
mad_perl_gene <- apply(exprSet, 1, mad, na.rm = TRUE)
cv_per_gene <- data.frame(mean = mean_per_gene,
sd = sd_per_gene,
mad=mad_perl_gene,
cv = sd_per_gene/mean_per_gene)
rownames(cv_per_gene) <- rownames(exprSet)
pairs(cv_per_gene)
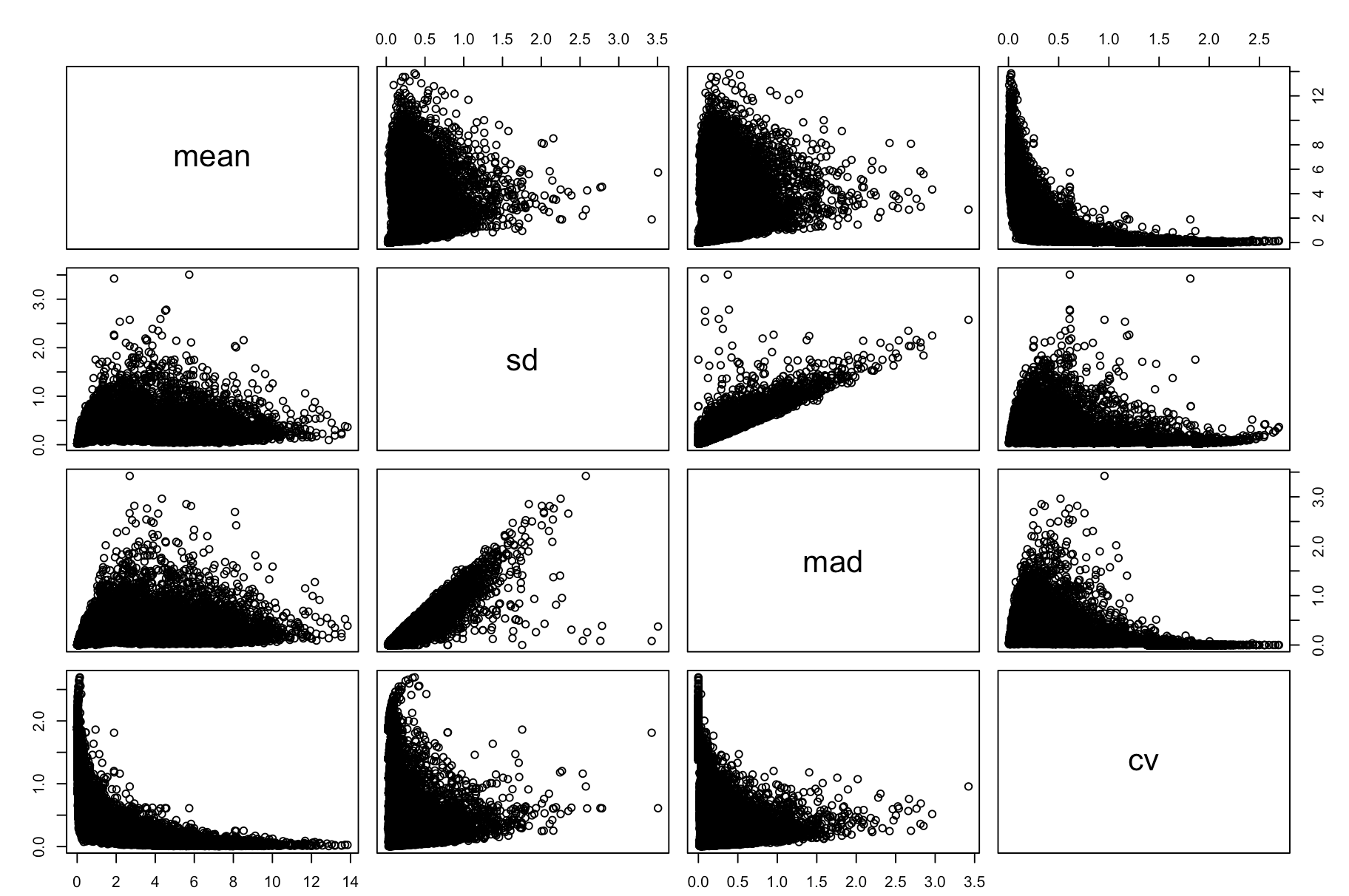
很明显,这个CV可以衡量某基因的表达变化情况,但是没办法在基因与基因之间比较,因为不同基因的CV不同,大部分情况是因为它们的平均表达量不同而已。
根据表达量对CV值进行校正
# https://jdblischak.github.io/singleCellSeq/analysis/cv-adjusted-wo-19098-r2.html
library(zoo)
# Compute a data-wide coefficient of variation on CPM normalized counts.
cv <- apply(2^exprSet, 1, sd)/apply(2^exprSet, 1, mean)
# Order of genes by mean expression levels
order_gene <- order(apply(2^exprSet, 1, mean))
# Rolling medians of log10 squared CV by mean expression levels
roll_medians <- rollapply(log10(cv^2)[order_gene], width = 50, by = 25,
FUN = median, fill = list("extend", "extend", "NA") )
## then change the NA values in the roll_medians
table(is.na(roll_medians))
ii_na <- which( is.na(roll_medians) )
roll_medians[ii_na] <- median( log10(cv^2)[order_gene][ii_na] )
names(roll_medians) <- rownames(exprSet)[order_gene]
# re-order rolling medians according to the expression matrix
roll_medians <- roll_medians[ match(rownames(exprSet), names(roll_medians) ) ]
stopifnot( all.equal(names(roll_medians), rownames(exprSet) ) )
# adjusted coefficient of variation on log10 scale
log10cv2_adj <- log10( cv^2) - roll_medians
plot(log10cv2_adj,mean_per_gene)
#install.packages("basicTrendline")
library(basicTrendline)
trendline(log10cv2_adj,mean_per_gene,model="line2P")
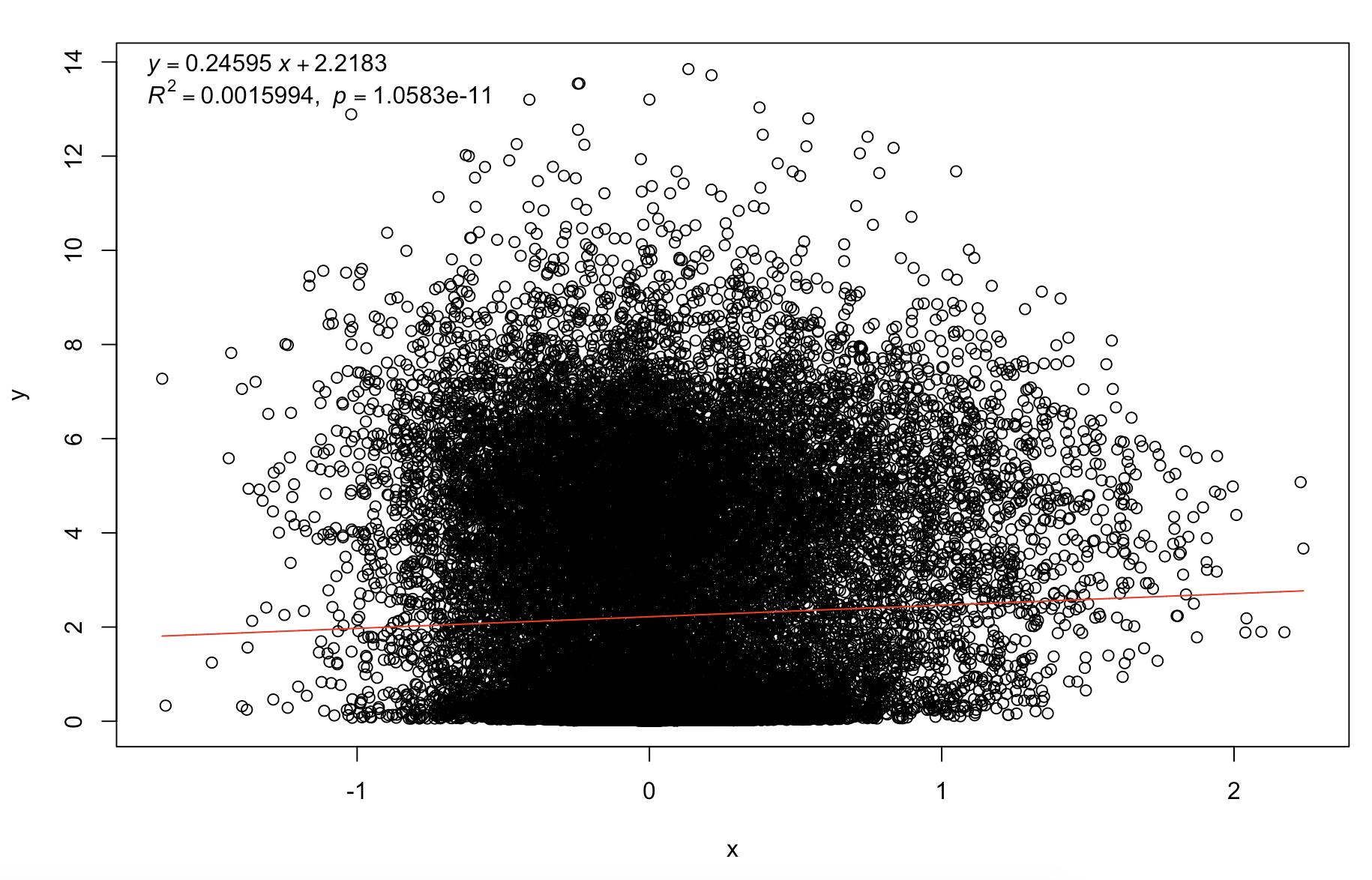
可以看到这个校正后的cv已经是几乎不受基因表达量的影响了,所以可以比较不同基因的表达变化情况啦。
根据基因长度对CV进行校正
先去gencode数据库找到gtf文件,对每个基因计算外显子长度之和作为基因的长度,代码如下;
## First, wecomputed gene lengths by taking the union of all exons within a gene based on the Ensembl annotation.
# cat ~/reference/gtf/gencode/gencode.v25.annotation.gtf|grep -v PAR_Y |perl -alne '{next if /^#/;if($F[2] eq "gene"){/(ENSG\d+)/;print $1} }'|sort |uniq -c |grep -w 2
# cat ~/reference/gtf/gencode/gencode.v25.annotation.gtf |grep -v PAR_Y |perl -alne '{next if /^#/;if($F[2] eq "gene"){/(ENSG\d+)/;$gene=$1;undef %h} if($F[2] eq "exon"){$key="$F[3]\t$F[4]";$len=$F[4]-$F[3];$c{$gene}+=$len unless exists $h{$key};$h{$key}++} }END{print "$_\t$c{$_}" foreach keys %c}' >>human_ENSG_length
# grep ENSG00000237094 ~/reference/gtf/gencode/gencode.v25.annotation.gtf|grep -w exon |cut -f 4-5|sort -u |awk '{print $2-$1}'|paste -sd+ - | bc
# grep ENSG00000237094 human_ENSG_length
# cat gencode.vM12.annotation.gtf |perl -alne '{next if /^#/;if($F[2] eq "gene"){/(ENSMUSG\d+)/;$gene=$1;undef %h} if($F[2] eq "exon"){$key="$F[3]\t$F[4]";$len=$F[4]-$F[3];$c{$gene}+=$len unless exists $h{$key};$h{$key}++} }END{print "$_\t$c{$_}" foreach keys %c}' >mouse_ENSG_length
得到的长度文件如下:
V1 V2 1 ENSG00000252040 131 2 ENSG00000251770 82 3 ENSG00000261028 856 4 ENSG00000186844 421 5 ENSG00000234241 1682 6 ENSG00000144815 15589
gen_l=read.table('human_ENSG_length',stringsAsFactors = F)
head(gen_l)
length_per_gene=gen_l[match(rownames(exprSet),gen_l[,1]),2]
mean_per_gene <- apply(exprSet, 1, mean, na.rm = TRUE)
sd_per_gene <- apply(exprSet, 1, sd, na.rm = TRUE)
mad_perl_gene <- apply(exprSet, 1, mad, na.rm = TRUE)
cv_per_gene <- data.frame(mean = mean_per_gene,
sd = sd_per_gene,
mad=mad_perl_gene,
len=log2(length_per_gene),
cv = sd_per_gene/mean_per_gene)
rownames(cv_per_gene) <- rownames(exprSet)
cv_per_gene=na.omit(cv_per_gene)
cor(cv_per_gene)
pairs(cv_per_gene)
fivenum(cv_per_gene$mean)
## 假如去除低表达量基因
cv_per_gene=cv_per_gene[cv_per_gene$mean < fivenum(cv_per_gene$mean)[2],]
pairs(cv_per_gene)
出图如下:
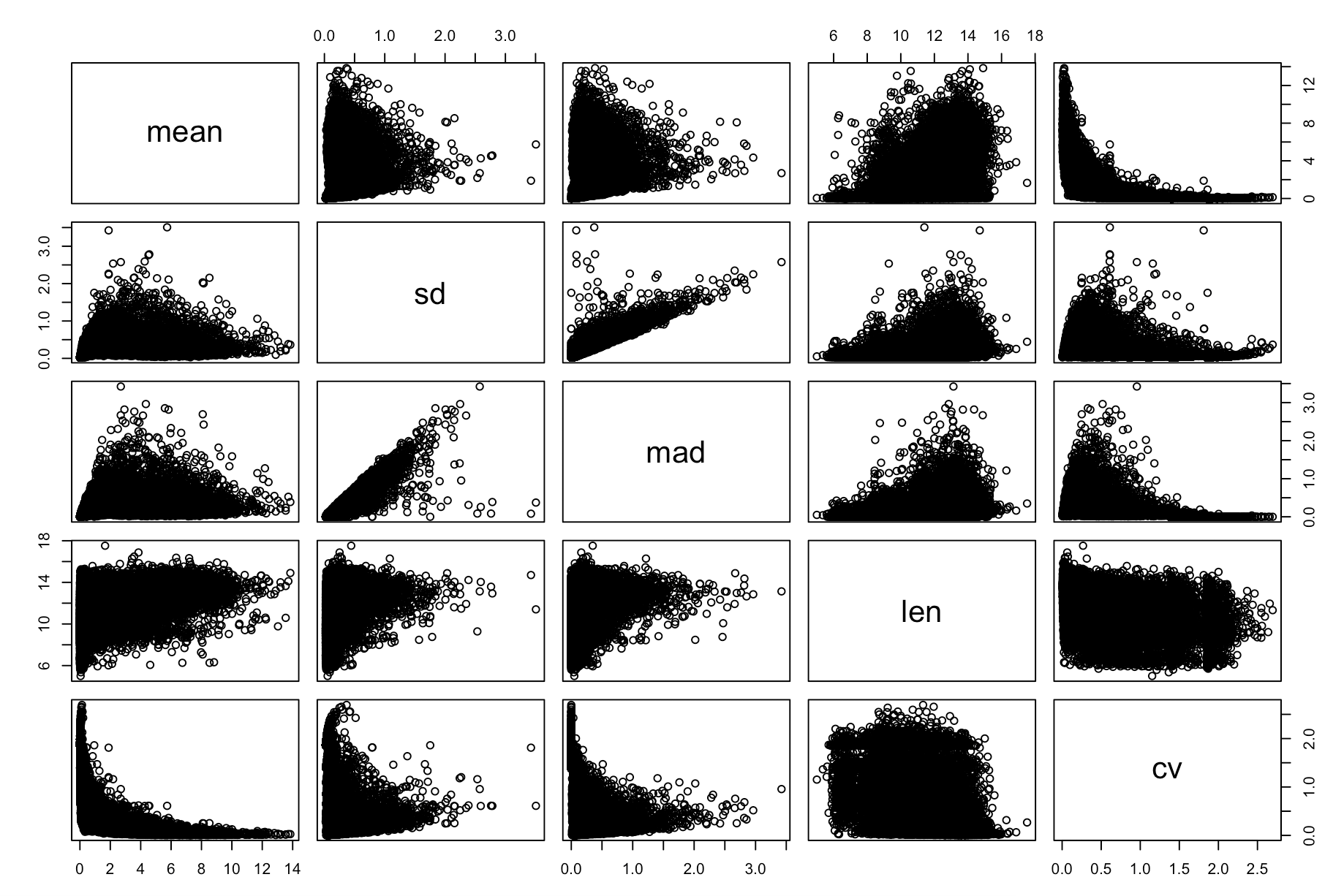
可以看到基因长度的确是影响着CV值,而且并不独立于表达量,所以还是需要去除这个因素。
可以使用校正表达量的代码来校正长度:
library(zoo)
table(rownames(exprSet) %in% gen_l[,1])
exprSet=exprSet[rownames(exprSet) %in% gen_l[,1],]
cv <- apply(2^exprSet, 1, sd)/apply(2^exprSet, 1, mean)
## firstly for mean values of exprSet
order_gene <- order(apply(2^exprSet, 1, mean))
roll_medians_mean <- rollapply(log10(cv^2)[order_gene], width = 50, by = 25,
FUN = median, fill = list("extend", "extend", "NA") )
## then change the NA values in the roll_medians_mean
table(is.na(roll_medians_mean))
ii_na <- which( is.na(roll_medians_mean) )
roll_medians_mean[ii_na] <- median( log10(cv^2)[order_gene][ii_na] )
names(roll_medians_mean) <- rownames(exprSet)[order_gene]
roll_medians_mean <- roll_medians_mean[ match(rownames(exprSet), names(roll_medians_mean) ) ]
stopifnot( all.equal(names(roll_medians_mean), rownames(exprSet) ) )
log10cv2_adj <- log10( cv^2) - roll_medians_mean
mean_per_gene <- apply(exprSet, 1, mean, na.rm = TRUE)
plot( log10( cv^2) ,mean_per_gene)
plot(log10cv2_adj,mean_per_gene)
length_per_gene=gen_l[match(rownames(exprSet),gen_l[,1]),2]
## Then for gene length(log10)
order_gene <- order( log10(length_per_gene) )
cv=log10cv2_adj
roll_medians_length <- rollapply(cv[order_gene], width = 50, by = 25,
FUN = median, fill = list("extend", "extend", "NA") )
## then change the NA values in the roll_medians_length
table(is.na(roll_medians_length))
ii_na <- which( is.na(roll_medians_length) )
roll_medians_length[ii_na] <- median( cv[order_gene][ii_na] )
names(roll_medians_length) <- rownames(exprSet)[order_gene]
roll_medians_length <- roll_medians_length[ match(rownames(exprSet), names(roll_medians_length) ) ]
stopifnot( all.equal(names(roll_medians_length), rownames(exprSet) ) )
log10cv2_adj <- cv - roll_medians_length
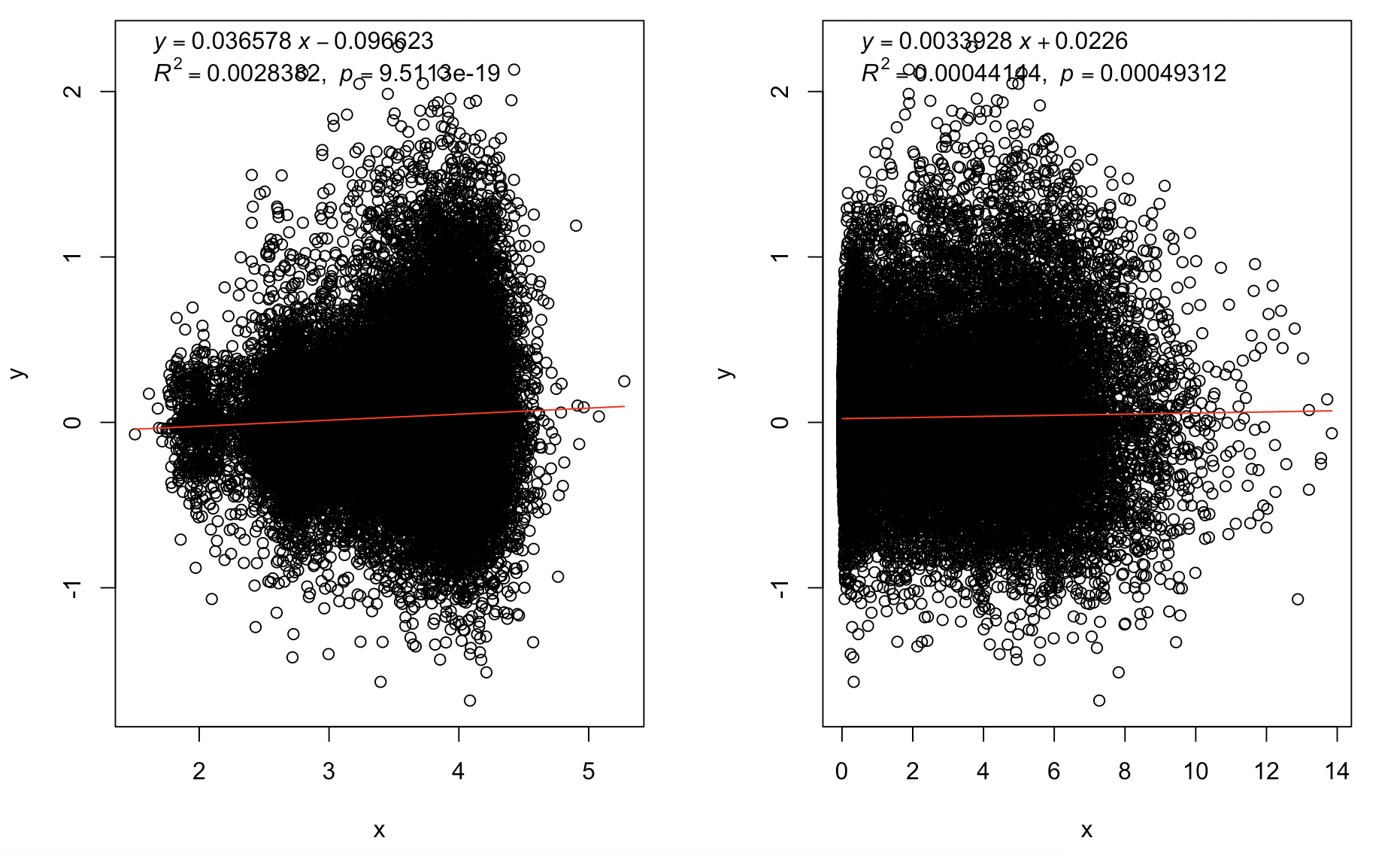
plot( log10( cv^2) ,log10(length_per_gene))
plot(log10cv2_adj,log10(length_per_gene))
plot(log10cv2_adj,log10(mean_per_gene))
#install.packages("basicTrendline")
library(basicTrendline)
par(mfrow=c(1,2))
trendline(log10(length_per_gene),log10cv2_adj,model="line2P")
trendline(mean_per_gene,log10cv2_adj,model="line2P")