Best practice for mRNA microarray
文章代码在:https://github.com/jmzeng1314/GEO
Note : Please don't use it if you are not the fan of our biotrainee, Thanks.
Install required packages by the codes below:
source("http://bioconductor.org/biocLite.R")
install.packages('devtools')
BiocInstaller::biocLite("jmzeng1314/biotrainee")
library(biotrainee)
But if you are in China, you should use the codes below:
install.packages("devtools", repos="https://mirrors.tuna.tsinghua.edu.cn/CRAN/")
library(devtools)
source("https://bioconductor.org/biocLite.R")
options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/")
BiocInstaller::biocLite('org.Hs.eg.db')
install.packages("remotes",repos="https://mirror.lzu.edu.cn/CRAN/")
BiocInstaller::biocLite("jmzeng1314/biotrainee")
install.packages("pheatmap",repos="https://mirror.lzu.edu.cn/CRAN/")
It will install many other packages for you automately, such as : ALL, CLL, pasilla, airway ,limma,DESeq2,clusterProfiler , that's why it will take a long time to finish if all of these packages are not installed before in your computer.
Then run step1 :
It always not very easy to download data if you are in China, so I also upload the file GSE42872_raw_exprSet.Rdata , you can load it directly.
if(F){
library(GEOquery)
gset <- getGEO('GSE42872', destdir=".",
AnnotGPL = F,
getGPL = F)
save(gset,'GSE42872.gset.Rdata')
}
load('GSE42872_eSet.Rdata')
b = eSet[[1]]
raw_exprSet=exprs(b)
group_list=c(rep('control',3),rep('case',3))
save(raw_exprSet,group_list,
file='GSE42872_raw_exprSet.Rdata')
Then step2:
Try to understand my codes, how did I filter the probes by the annotation of each microarry, and how I check the group information for the different samples in each experiment.
Including PCA and Cluster figures, as below:


Please ensure that you do run those codes by yourself !!!
Then step3:
Normally we will do differential expression analysis for the microarray, and LIMMA is one of the best method, so I just use it. If the expression matrix(raw counts ) comes from mRNA-seq, you can also choose DESeq based on negative binomial (NB) distributions or baySeq and EBSeq.
Once DEG finished, we can choose top N genes for heatmap as below:

and volcano plot as below:
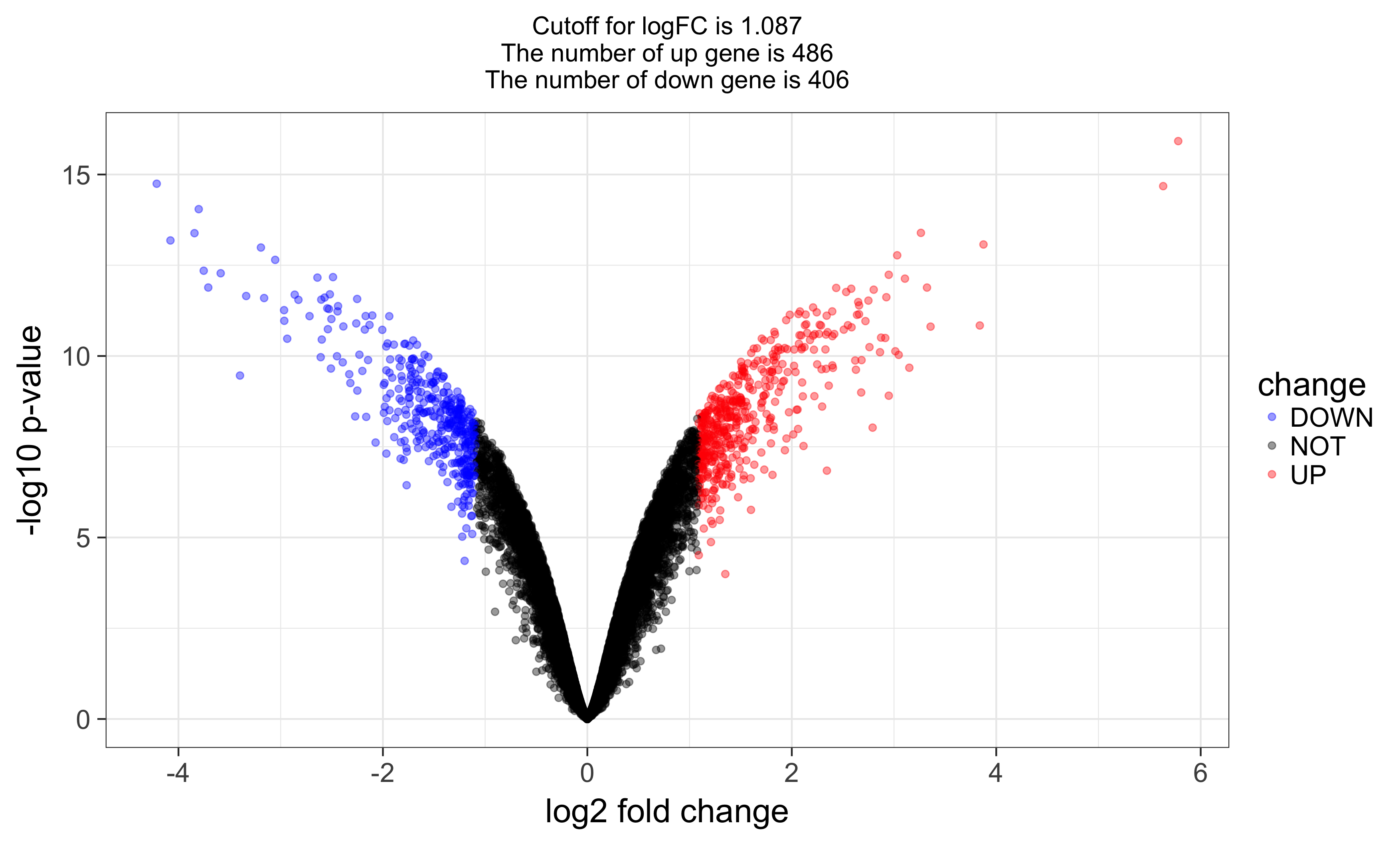
Last step :
Annotation for the significantly changed genes, over-representation test or GSEA for GO/KEGG/biocarta/rectome/MsigDB and so on.


最重要的是:
如果你觉得我的教程对你有帮助,请赞赏一杯咖啡哦!
如果你的赞赏超过了50元,请在扫描赞赏的同时留下你的邮箱地址,我会发送给你一个惊喜哦!
