前面我们分享的GEO数据库挖掘教程都是针对表达芯片来的,会给粉丝们一种错觉,是不是这个技术只能挖掘这些老旧的表达芯片呢?
当然不是这样,现在就给大家秀一秀RNA-seq数据的挖掘。
往期目录:
GEO数据挖掘系列文-第一期-胶质母细胞瘤
GEO数据挖掘系列文-第二期-三阴性乳腺癌
GEO数据挖掘系列文-第三期-口腔鳞状细胞癌
GEO数据挖掘系列文-第四期-肝细胞癌 (WGCNA)
GEO数据挖掘系列文-第五期-肝细胞癌 (多组差异分析)
外传:保姆式GEO数据挖掘演示—重现9分文章
文章解读
具体文章的生物学故事及背景,大家可以看我的文献分享:https://zhuanlan.zhihu.com/p/49574925 第47周-多组学探索不同器官的小细胞癌症起源
这里就看看测序方面,而且是RNA-seq的,如下:

既然给出了测序数据,那么我们就可以完完全全的重复该流程。
首先进入GEO数据库找到它:
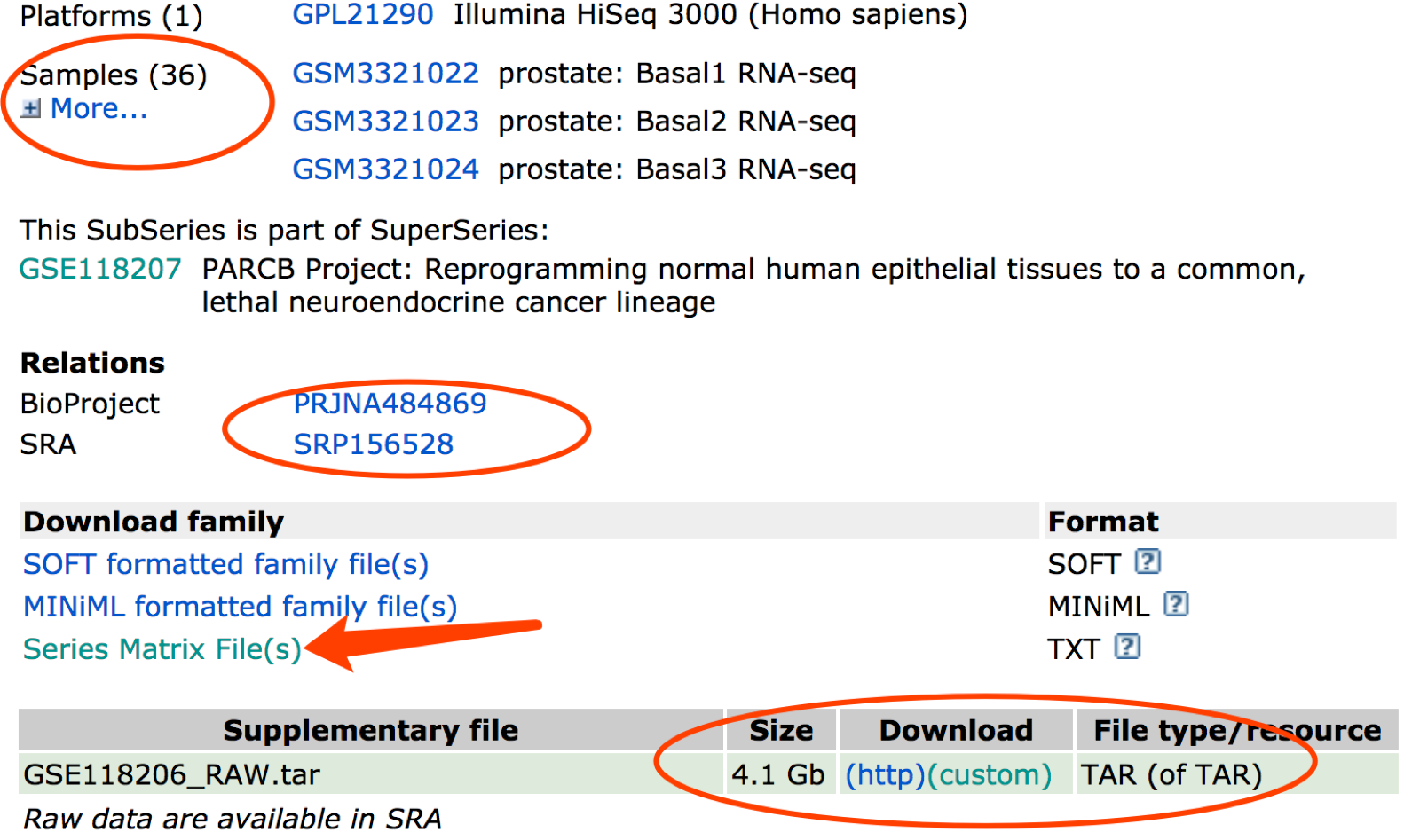
仅仅是信号bw格式文件都是4.1Gb了,而且作者没有提供表达矩阵供我们下载,所以我们需要自行下载测序数据;
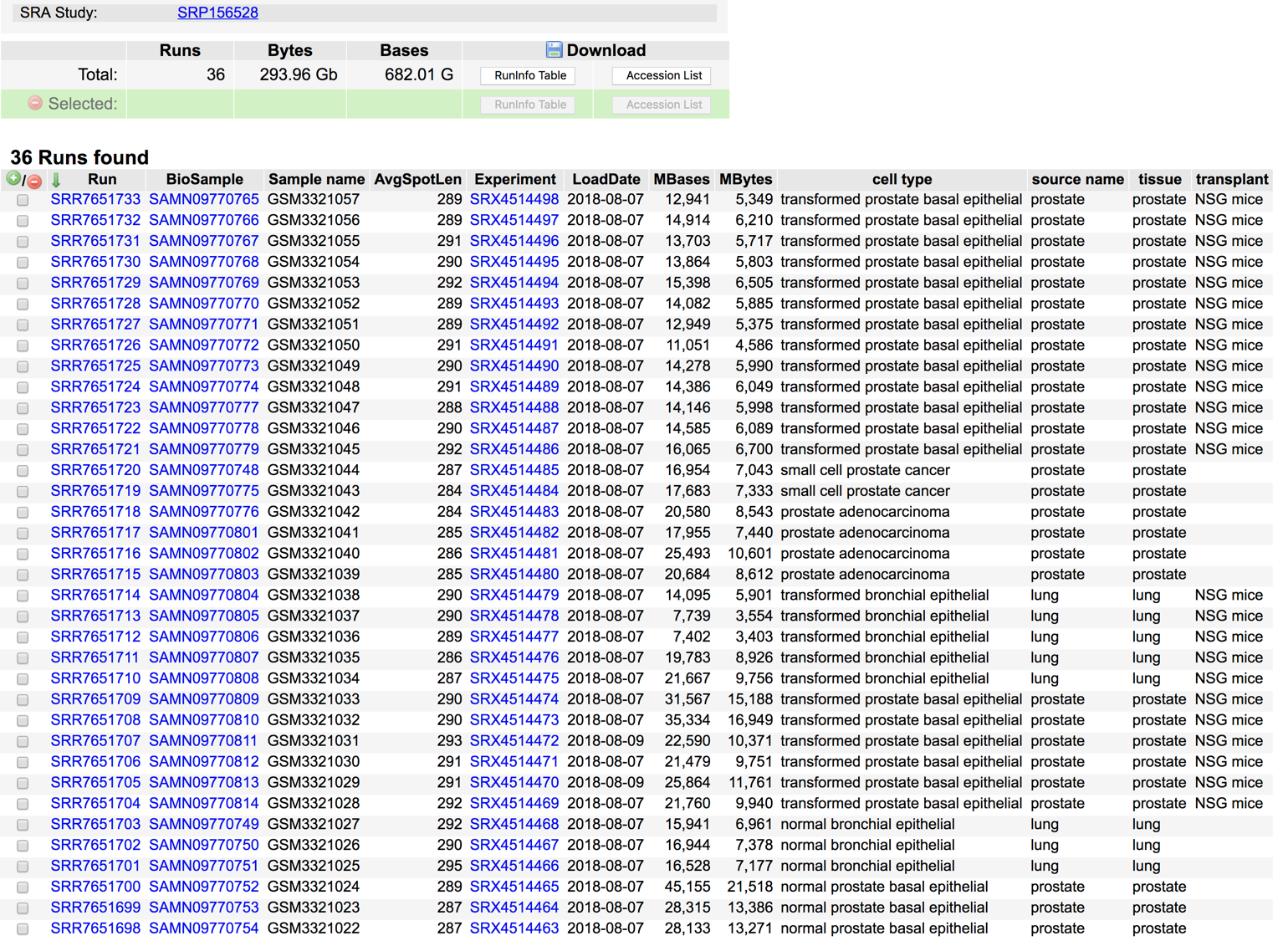
数据量不小,按照我在生信技能树的教程,首先应该是学习了解GEO和SRA数据库:
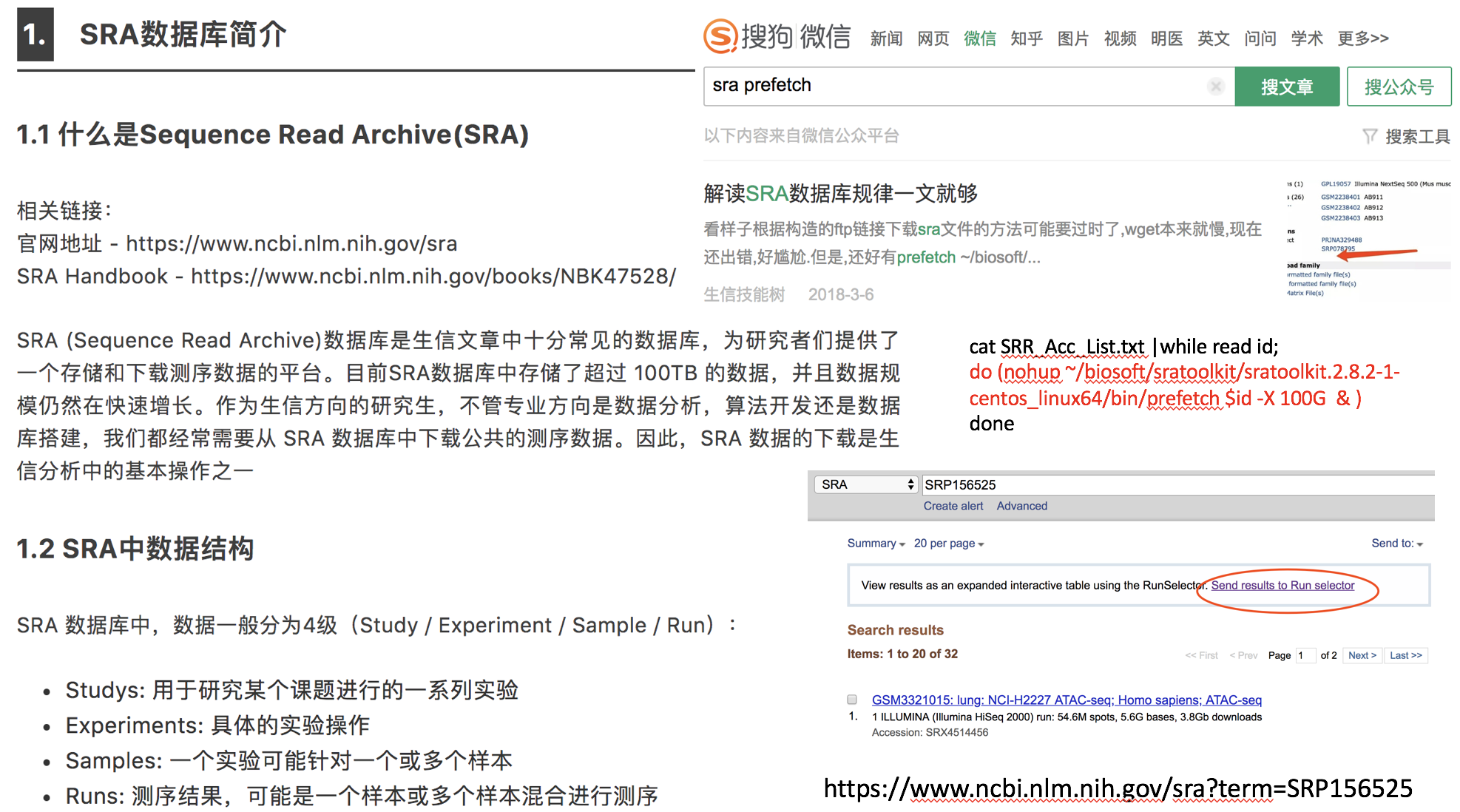
然后应该是下载sra-toolkit软件来下载原始数据:
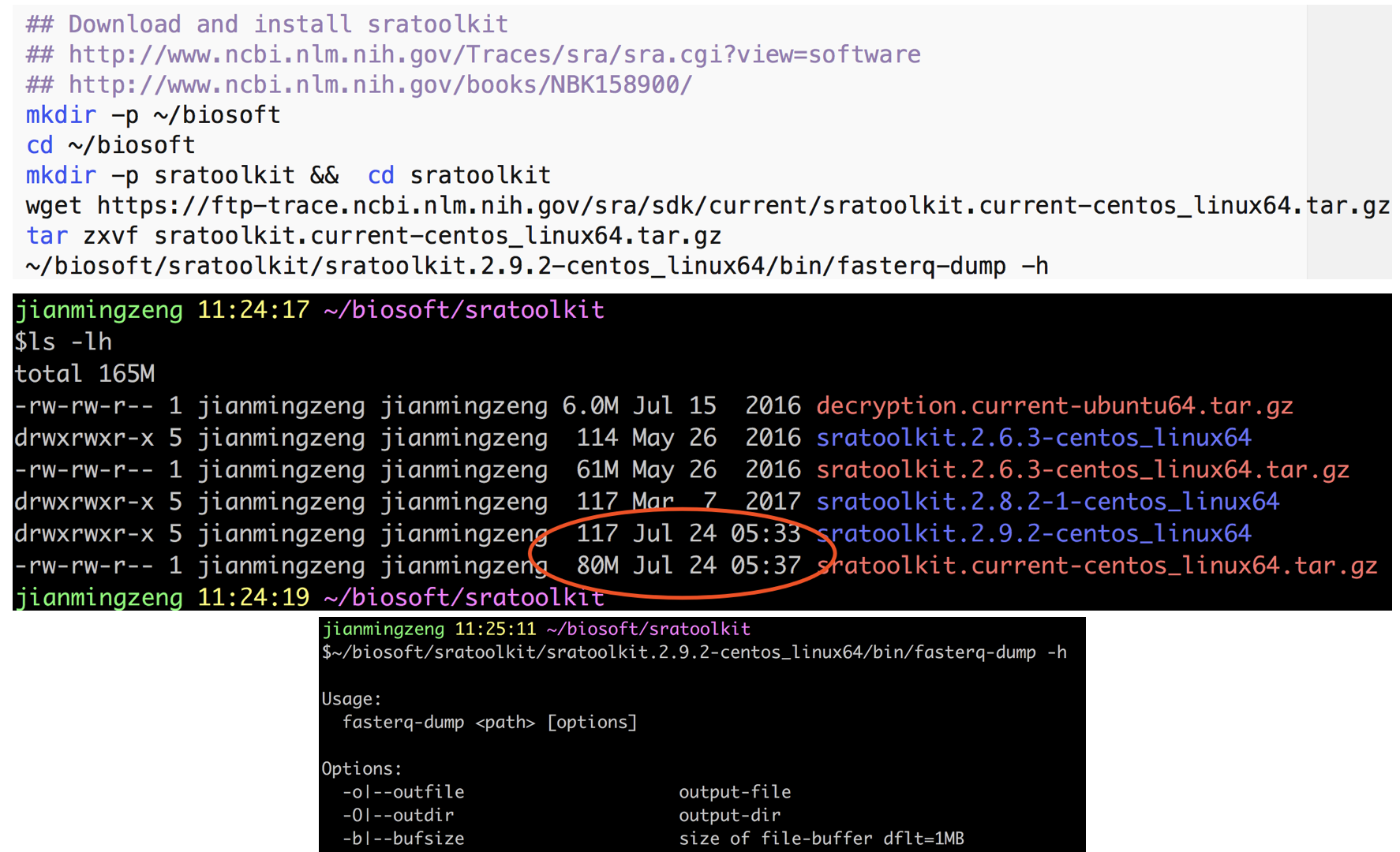
有了工具,下载无非就是一个命令行而已,数据比较大,可能是会比较耗时。
sra格式的测序数据需要转为fastq格式,然后需要定表达量,可以直接使用salmon这样的不需要比对就能定量的工具,考虑到很多读者并不一定能看懂shell代码,我这里就不列出了,感兴趣可以直接去生信技能树的B站免费学习这些知识。
表达矩阵重现
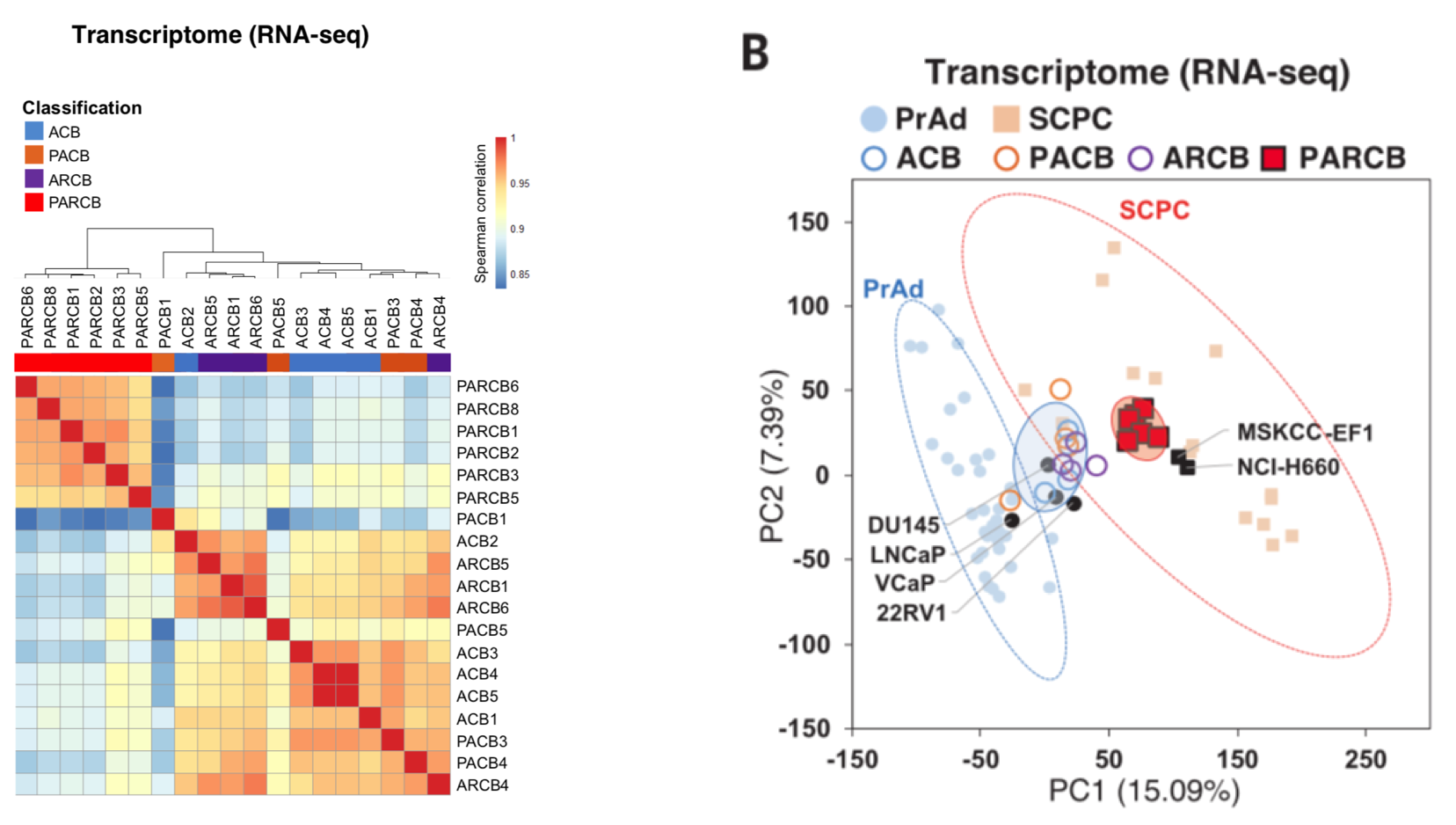
经过不懈努力,我使用salmon得到了表达矩阵,然后就能导入R进行下游分析,先从这个聚类关系图开始吧。
代码是:
rm(list=ls())
options(stringsAsFactors = F)
load('quants-exprSet.Rdata') ## 这个数据集来源于 salmon得到了表达矩阵
# 如果需要重现这个流程,可以发邮件找我(jmzeng1314@163.com)申请这个数据集:quants-exprSet.Rdata
exprSet[1:4,1:4]
dim(exprSet)
colnames(exprSet)
exprSet=exprSet[,grepl('CB',colnames(exprSet))]
exprSet=exprSet[,!grepl('NHBE',colnames(exprSet))]
group_list=gsub('[1-9]$','',colnames(exprSet))
dat=t(exprSet)
dat=as.data.frame(dat)
dat=cbind(dat,group_list)
# Define nodePar
nodePar <- list(lab.cex = 0.6, pch = c(NA, 19) ,
cex = 0.4, col = "blue")
hc=hclust(dist(t(log(edgeR::cpm(exprSet)+1))))
par(oma=c(5,5,5,30))
png('hclust.png',res=120,height = 680,width = 480)
hcd=as.dendrogram(hc)
# Horizontal plot
plot(hcd, xlab = "Height",
nodePar = nodePar, horiz = TRUE)
dev.off()
## PCA
library("FactoMineR")
library("factoextra")
# The variable group_list (index = 54676) is removed
# before PCA analysis
dat.pca <- PCA(dat[,-ncol(dat)], graph = FALSE)
fviz_pca_ind(dat.pca,
geom.ind = "point", # show points only (nbut not "text")
col.ind = dat$group_list, # color by groups
#palette = c("#00AFBB", "#E7B800"),
addEllipses = TRUE, # Concentration ellipses
legend.title = "Groups"
)
由上面的代码,首先出图:

由于作者只考虑PARCB这5个基因的分组情况的聚类,所以我们挑选适合的表达矩阵进行下游PCA分析看远近关系如下:
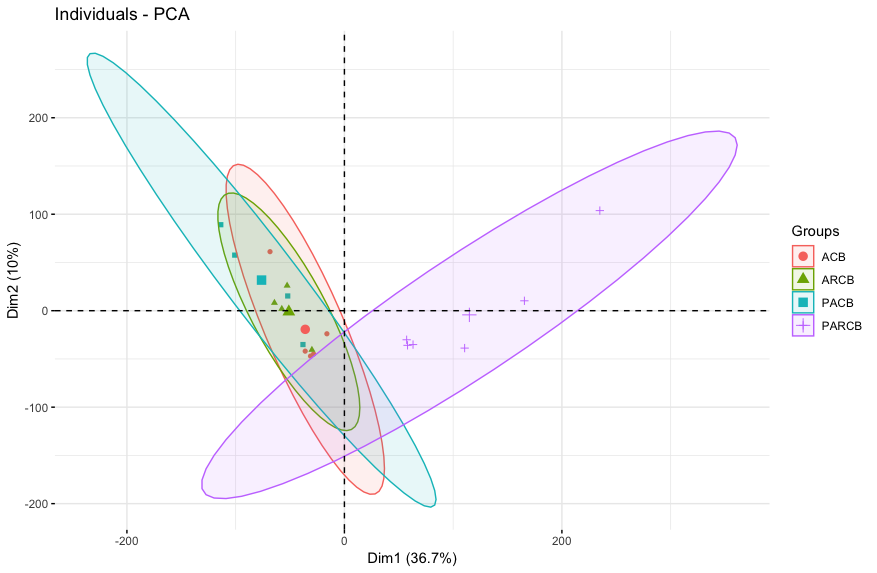
和原文作者的相关性热图结论一致,都是PARCB这个组别要显著与其它组分离。