你是否害怕linux的黑白命令行操作,是否对去可视化畏畏缩缩,那么你会爱上它:Rsubread这里演示一下传统的RNA-seq数据的表达量分析全流程, 安装Rsubread包后会有自带的测序数据如下:
-rw-r--r-- 1 jmzeng admin 25K Nov 9 18:04 longreads.txt.gz
-rw-r--r-- 1 jmzeng admin 80K Nov 9 18:04 reads.txt.gz
-rw-r--r-- 1 jmzeng admin 80K Nov 9 18:04 reads1.txt.gz
-rw-r--r-- 1 jmzeng admin 80K Nov 9 18:04 reads2.txt.gz
-rw-r--r-- 1 jmzeng admin 89K Nov 9 18:04 reference.fa
下面的分析流程也以此为例子,不过要切记,一旦切换到人类真实数据,下面的步骤都会耗时很可观,要有心理准备哈!
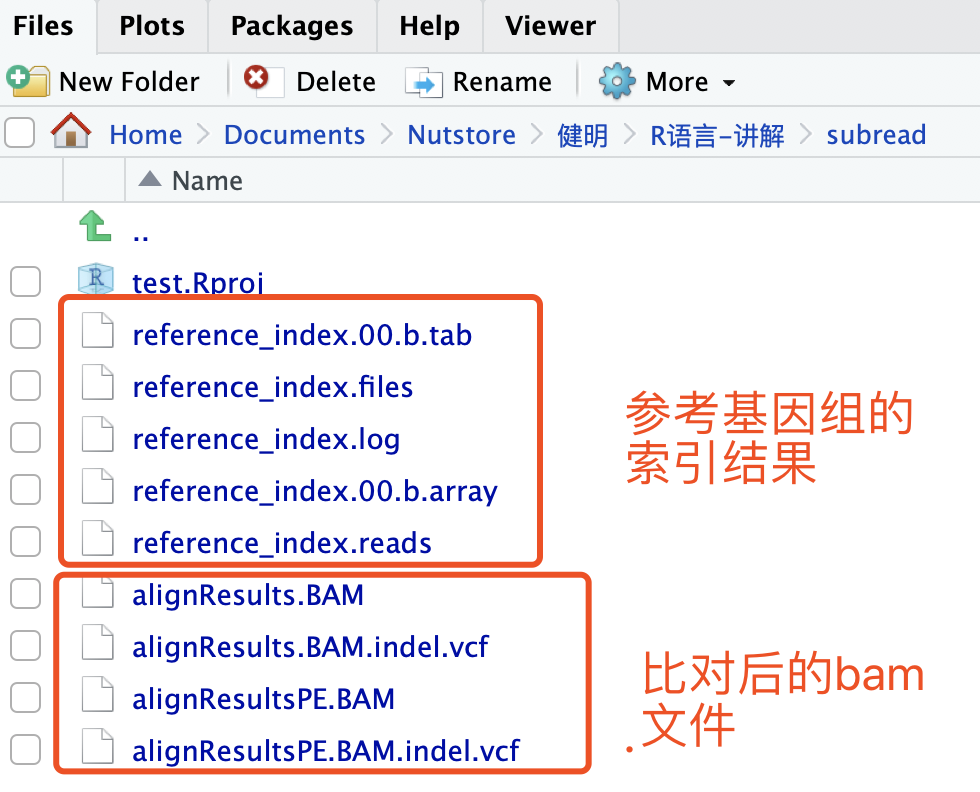
step1:构建索引
需要有参考基因组文件,这里使用Rsubread自带的数据作为例子:
library(Rsubread)
ref <- system.file("extdata","reference.fa",package="Rsubread")
buildindex(basename="reference_index",reference=ref)
step2:比对
需要有fastq格式的测序数据,还是使用Rsubread自带的数据作为例子:
## 首先是单端数据
reads <- system.file("extdata","reads.txt.gz",package="Rsubread")
align(index="reference_index",readfile1=reads,output_file="alignResults.BAM",phredOffset=64)
## 下面是双端
reads1 <- system.file("extdata","reads1.txt.gz",package="Rsubread")
reads2 <- system.file("extdata","reads2.txt.gz",package="Rsubread")
align(index="reference_index",readfile1=reads1,readfile2=reads2,
output_file="alignResultsPE.BAM",phredOffset=64)
测试数据比对很迅速,也会同步输出bam文件到本地。
step3:定量
需要有基因组特征描述文件,通常是gtf格式的基因,转录本,外显子的染色体,起始终止坐标,这里还是使用测试数据,自己制作 特征描述文件如下:
ann <- data.frame(
GeneID=c("gene1","gene1","gene2","gene2"),
Chr="chr_dummy",
Start=c(100,1000,3000,5000),
End=c(500,1800,4000,5500),
Strand=c("+","+","-","-"),
stringsAsFactors=FALSE)
ann
fc_SE <- featureCounts("alignResults.BAM",annot.ext=ann)
fc_SE
fc_PE <- featureCounts("alignResultsPE.BAM",annot.ext=ann,isPairedEnd=TRUE)
fc_PE
是不是很激动,这么简单就完成了NGS组学数据分析一条龙分析啊!!!
还有一些小细节
x <- qualityScores(filename=reads,offset=64,nreads=1000)
x[1:10,1:10]
propmapped("alignResults.BAM")
值得注意的是,你只是看了看这个包的用法而已,要想用得好,请听下回分解哦!
推荐看原版50页的PDF说明书哈:https://bioconductor.org/packages/release/bioc/vignettes/Rsubread/inst/doc/SubreadUsersGuide.pdf