很久以前我们提到过TCGA的各种网页数据库的生存分析结果冲突的问题,现在又有人提出来一个新的问题,如下:
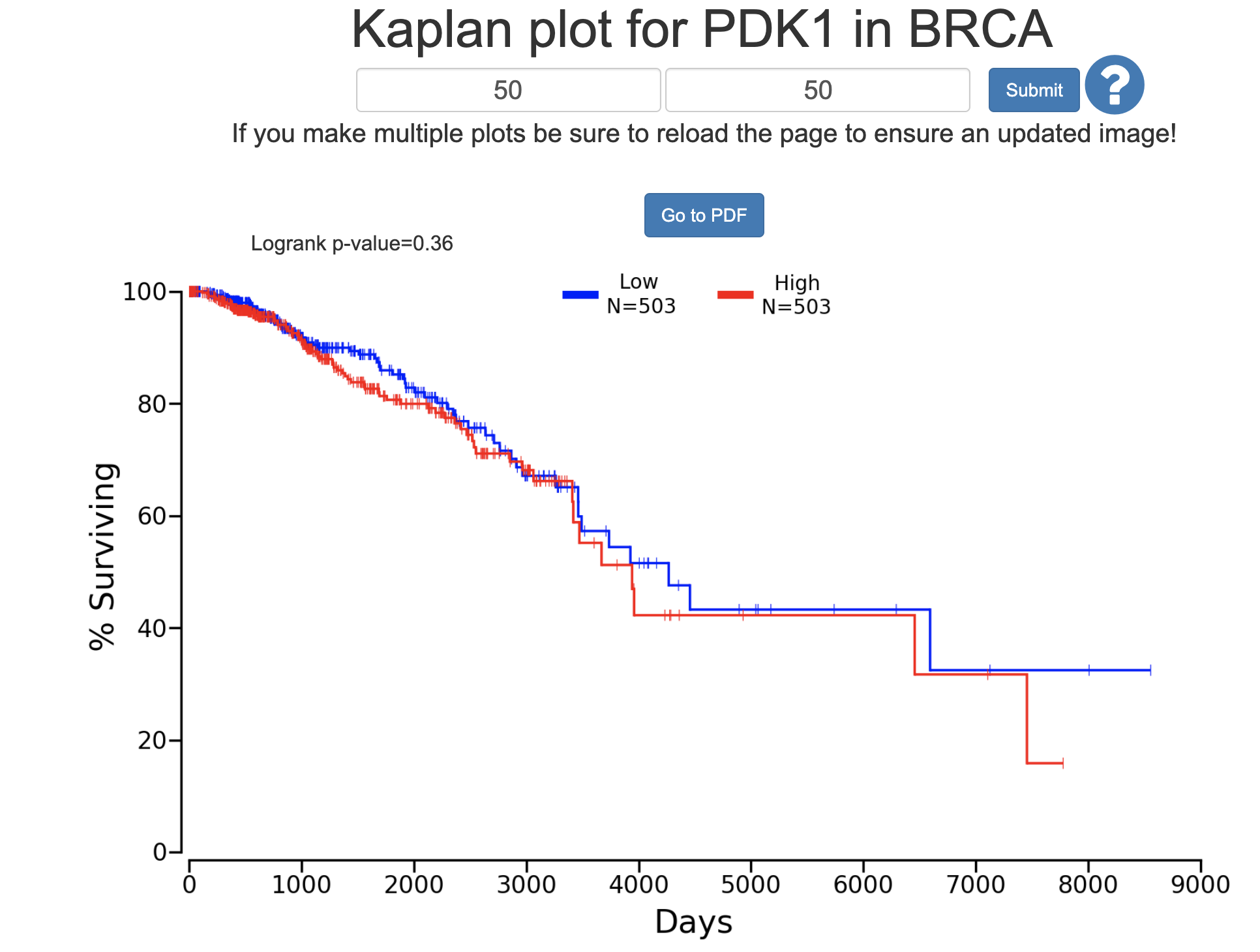
根据基因表达量的中位值把样本分成高低表达量的组别,然后做差异分析是比较符合大家的直觉的。
如果这个时候生存分析结果不具有统计学显著性,而大家又的确感兴趣这个基因在这个癌症的临床意义,会尝试调整分组指标,这也就是为什么网页工具会提供调整阈值的窗口,比如调整为如下所示:
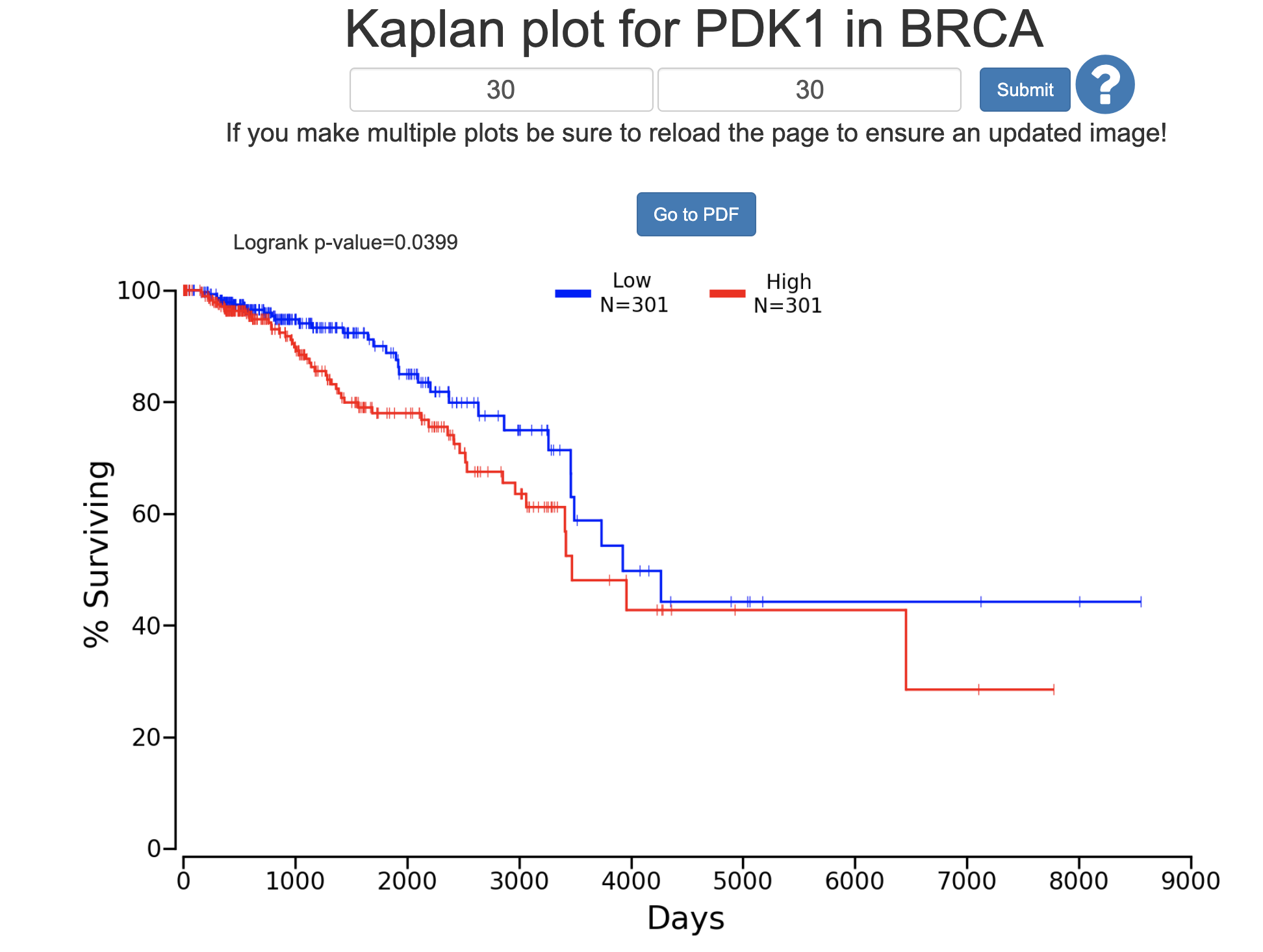
你会惊奇的发现,显著了!!!
但是实际上这样30%的阈值来进行分组的操作一定会受到审稿人质疑,基本上没有人这样操作,如果调整我25%的阈值就会发现马上又不显著了。
所以这样的KM分析是有弊端的!
那COX分析呢
COX分析就是排除一下样本其它信息的干扰之后的生存分析,这个时候网页工具能做的很有限,我们需要下载临床数据在R里面完成这个分析,如果你看了我的视频,就应该是知道至少下面两个临床信息是值得信赖的。
- https://gdc.xenahubs.net/download/TCGA-BRCA/Xena_Matrices/TCGA-BRCA.GDC_phenotype.tsv.gz
- TCGA-CDR https://api.gdc.cancer.gov/data/1b5f413e-a8d1-4d10-92eb-7c4ae739ed81
打开Rstudio,接下来就开始我们的表演吧!
首先制作网页工具同样的图:a=read.csv('BRCA_5163_50_50.csv') head(a) a$event=ifelse(a$Status=='Alive',0,1) library(survival) library(survminer) sfit <- survfit(Surv(Days, event)~Group, data=a) ggsurvplot(sfit, conf.int=F, pval=TRUE)出图如下:
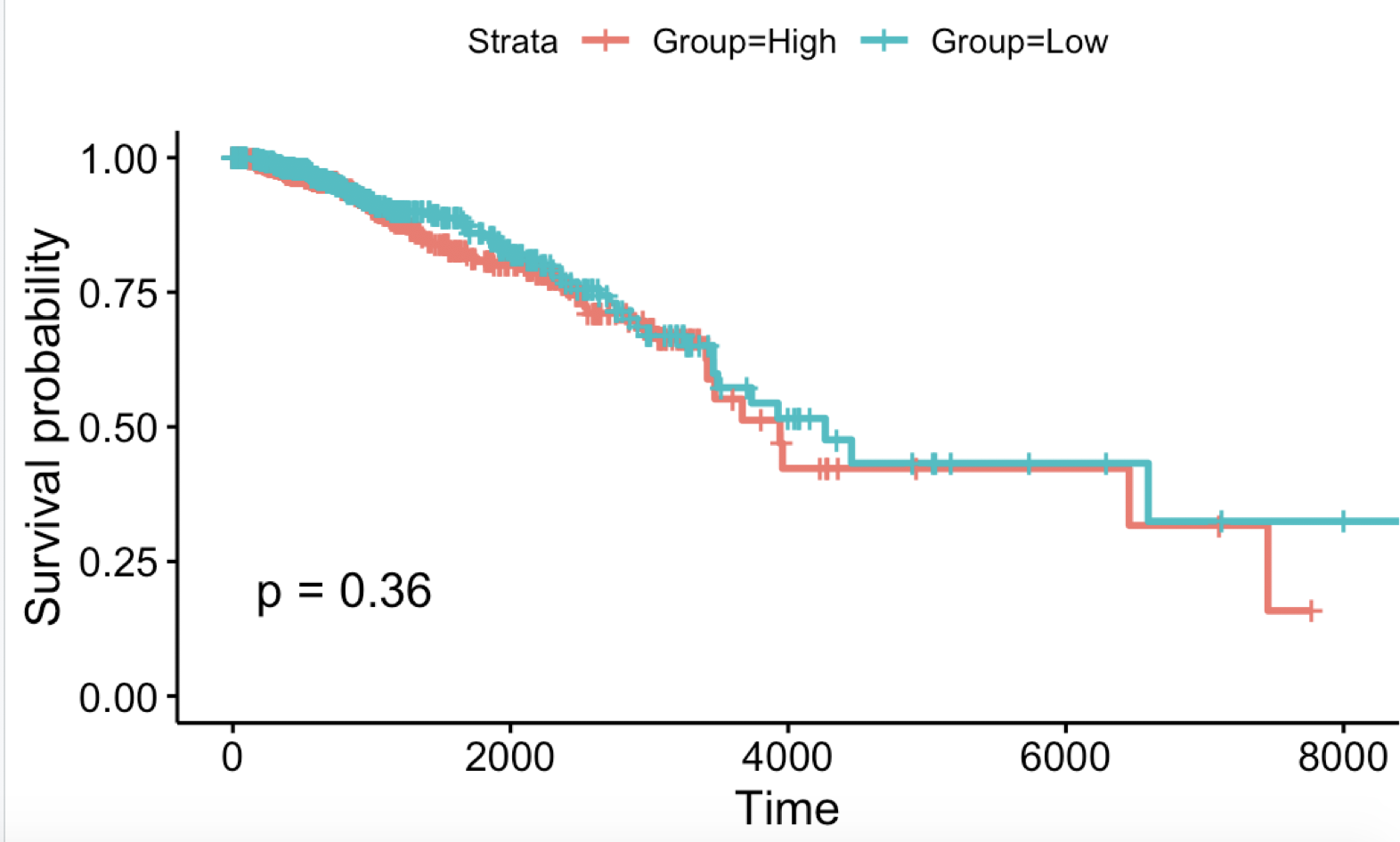
读者朋友们这里可以尝试写一个循环,看看不同阈值分组的生存分析的统计学P值如何。
接下来读入临床信息,做COX分析,这个时候需要对临床信息有非常深刻的认识,毕竟临床记录动辄都是几百种信息。
挑选几个自己觉得值得探索的因素去做COX分析,继续看不同分类标准后的基因表达量分组是否生存分析具有统计学显著性。
代码参考:https://github.com/jmzeng1314/tcga_example/blob/master/scripts/step04-batch-coxp.R那区分亚型呢
对BRCA的亚型,可以在 https://gdc.cancer.gov/about-data/publications/panimmune 里面下载到
文件是:Annotated TCGA Subtypes by Noushmehr and Malta - TCGASubtype.20170308.tsv - https://api.gdc.cancer.gov/data/0f31b768-7f67-4fc4-abc3-06ac5bd90bf0
那,有了这些文件,很容易写代码去探索不同亚型内部该基因是否具有KM或者COX的生存分析的统计学显著性。