文章发表于 Annals of Oncology, April 2018, 链接是,https://doi.org/10.1093/annonc/mdy024
主要是下载两个数据库总共 (n = 550) 的TNBC病人的3数据进行分析,即:
- Molecular Taxonomy of Breast Cancer International Consortium (METABRIC)
- The Cancer Genome Atlas. TNBC samples
这3种数据分别是: - copy-number aberrations
- somatic mutations
- gene expression
进行了整合分析,所谓的多组学整合,其实就是根据表达数据对样本进行分组, 然后看CNV和SNV层面的差异。关于TNBC分组背景
最开始2011的那篇meta分析把TNBC分成6类;
- basal-like 1 (BL1), basal-like 2 (BL2), immunomodulatory (IM), luminal androgen receptor (LAR), mesenchymal (M) and mesenchymal stem-like (MSL)
后来作者重新修正了,为4类: - basal-like/immune-suppressed (BLIS)
- basal-like/immune activated (BLIA)
- luminal androgen receptor (LAR)
- mesenchymal (MES)
关于TNBC分类问题,在2019年文献分享第09篇有详细整理介绍过,这里就不再赘述,我们直接来看看作者是如何整理不同数据库的TNBC病人的3种组学数据的。TNBC数据下载
在下面两个数据库,可以分别下载到总共550个病人数据,分别是355 and 195 TNBC samples
- Molecular Taxonomy of Breast Cancer International Consortium (METABRIC)
- The Cancer Genome Atlas Consortium (TCGA)
使用consensus聚类
这里直接可以使用 https://www.bioconductor.org/packages/release/bioc/html/ConsensusClusterPlus.html
主要是基于 cumulative distribution function (CDF) curve 选择合适的群,这里选择5群:
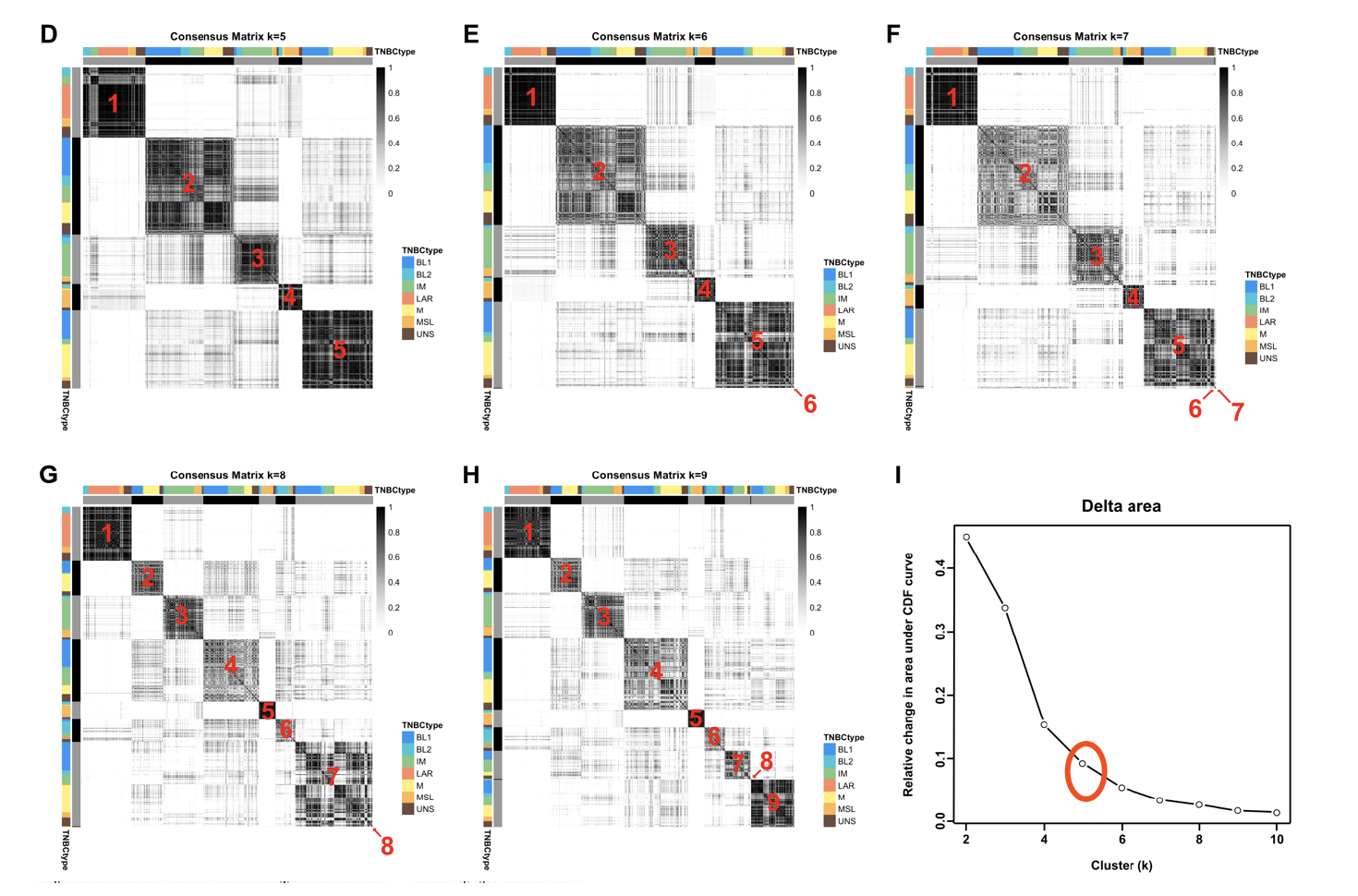
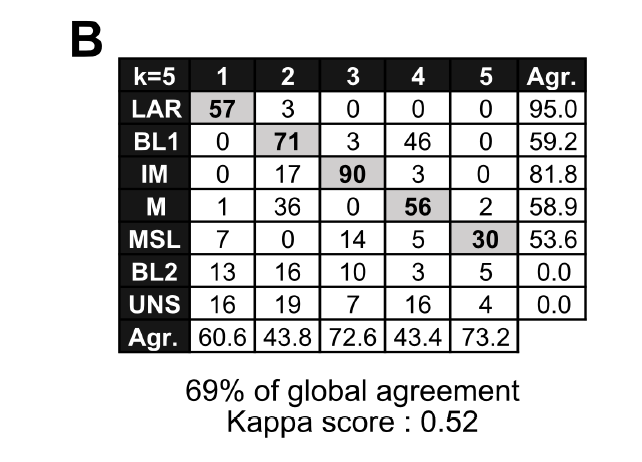
consensus聚类结果跟2011的 meta分类结果对比
可以看到全局overlap还行,这样作者自己的5群, 就可以根据overlap来进行命名啦。
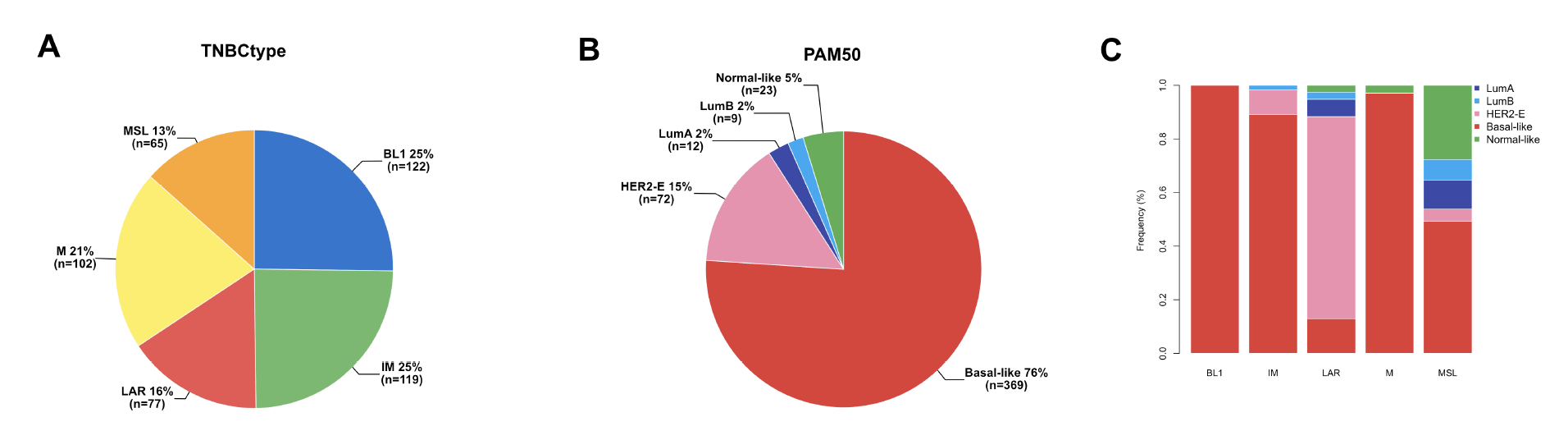
consensus聚类结果跟PAM50比较
可以看到,是符合认知的,大多数TNBC都是PAM50分类的basal-like亚型
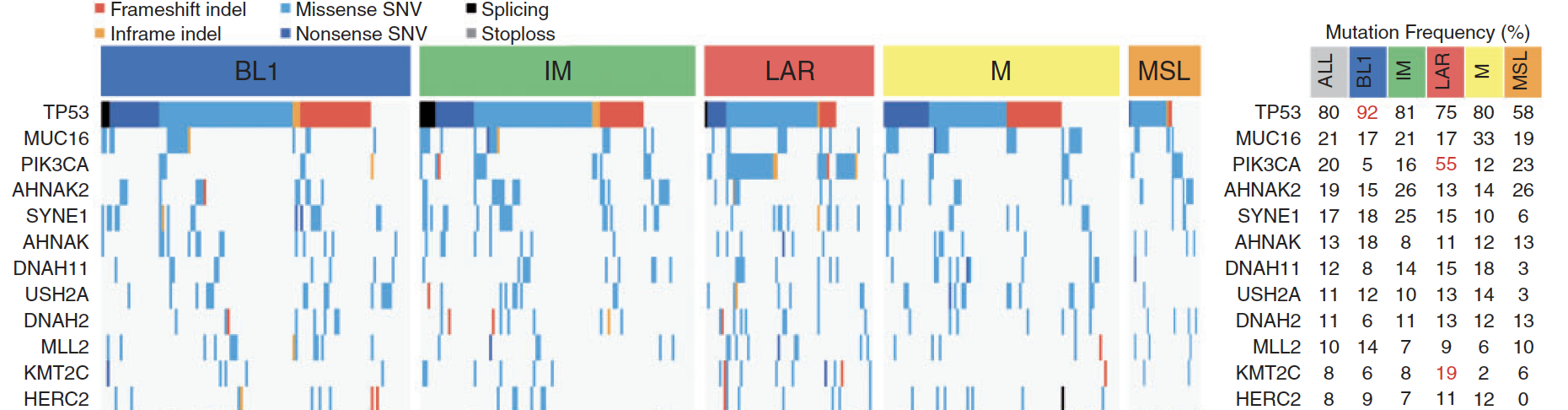
consensus分类后看突变差异
首先可以看突变全景图,这里作者就可以大片段描述
还可以看一些简单的统计指标 - Frequency distribution of substitution type,
- mutation effect on protein in the genes.
- Mutational burden for each TNBC molecular subtype considering all exonic mutations
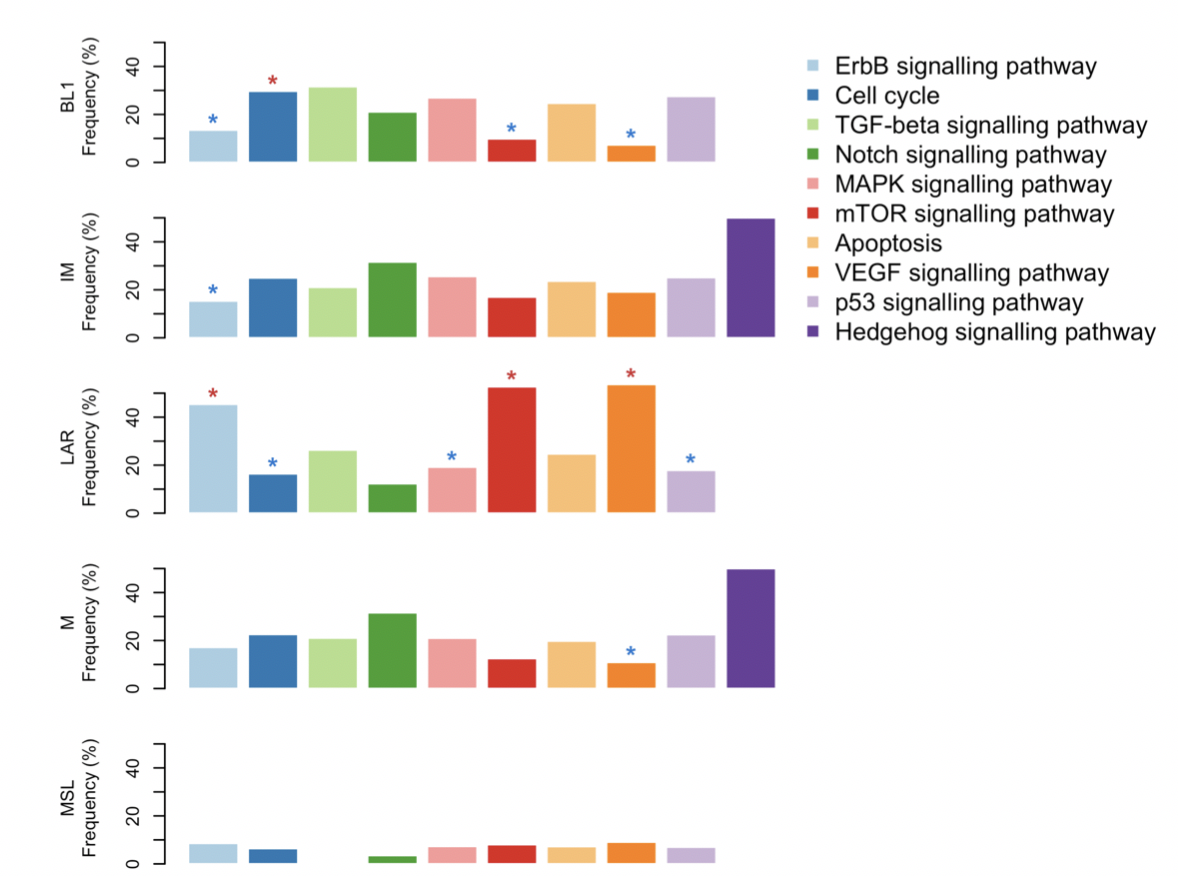
作者在这里展现了另外一种比较方式,不同亚型的不同pathway基因的突变频率。
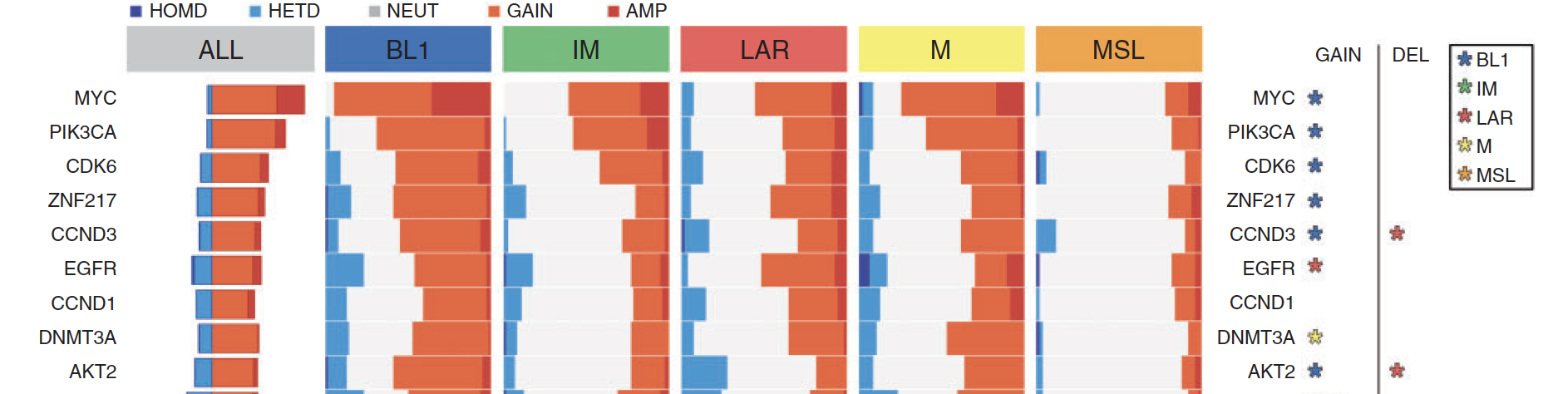
consensus分类后看基因组不稳定性
主要是拷贝数变异,同样是全景图展现:
然后使用 Chromosomal instability 值的分布情况来比较不同亚型的TNBC区别。
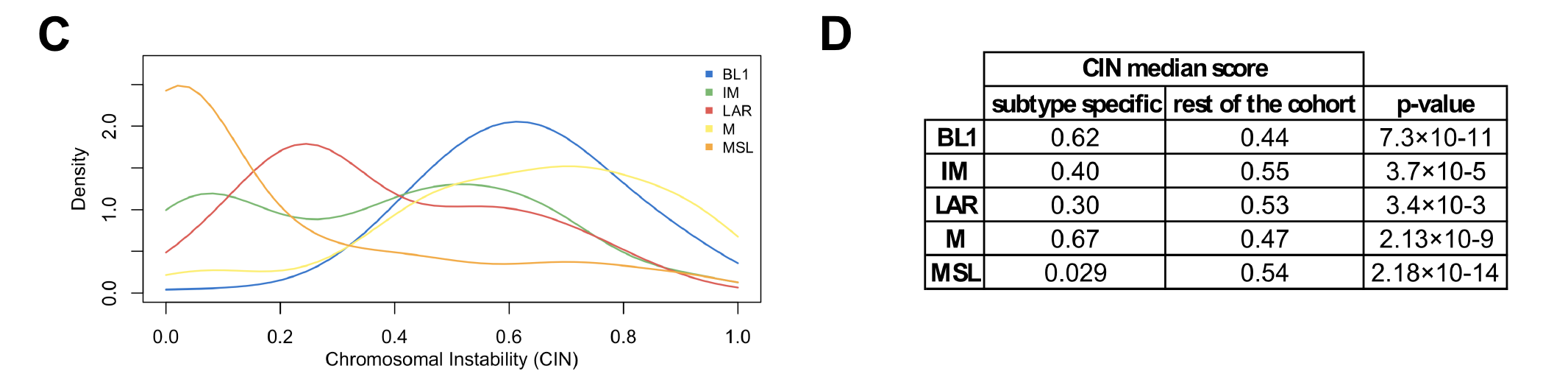
这里的chromosomal instability (CIN) 定义很简单,就是 the percentage of the genome affected by CNAs according to hormonal status后记
总体上来说,本文没有啥亮点,都是很常规的分析,就如同我开始评价的,所谓的多组学整合,其实就是根据表达数据对样本进行分组, 然后看CNV和SNV层面的差异。