最近学徒刚刚完成了RNA-seq的训练,我就随机抽取了一个公共数据库项目给他作为作业,研究者是通过CRISPR/Cas9对 nasopharyngeal carcinoma (NPC) 细胞系的p53引入了一个杂合突变 R280T ,然后看mRNA profiles of CNE2 (control) and KO CNE2 cells 的差异。
可以看到是6个样本:
| gse| description|
| —————————————————————————————— | ————- |
| GSM3737482 | CNE2(WTa) |
| GSM3737483 | CNE2(WTb) |
| GSM3737484 | CNE2(WTc) |
| GSM3737485 | CNE2-KO41a |
| GSM3737486 | CNE2-KO41b |
| GSM3737487 | CNE2-KO41c |
作者提供了表达矩阵:GSE130398_fpkm.txt.gz
第一个流程是 hisat2+featureCounts
第二个流程是salmon
第三个流程在文章里面
但是找不到文章,除非去询问作者,Xiangya Hospital, Central South University 的 zhenqi qin
首先查看突变是否引入成功
首先查看bam文件,头文件:
@PG ID:hisat2 PN:hisat2 VN:2.1.0 CL:"/project/home/lyang/miniconda2/bin/hisat2-align-s --wrapper basic-0 -p 10 -x /project/home/lyang/refdata/hisat/human/hg38/genome -S SRR8980083.sam -1 /tmp/3190.inpipe1 -2 /tmp/3190.inpipe2"
流程使用的是hg38参考基因组 , 简单搜索目标基因:https://www.ncbi.nlm.nih.gov/gene/7157
GRCh38.p13 (GCF_000001405.39) 17 NC_000017.11 (7668402..7687550, complement)
也可以在gtf文件搜索:
chr17 HAVANA gene 7661779 7687550 . - . gene_id "ENSG00000141510.17"; gene_type "protein_coding"; gene_name "TP53"; level 2; hgnc_id "HGNC:11998"; tag "overlapping_locus"; havana_gene "OTTHUMG00000162125.10";
批量提取 tp53 坐标的小bam:
ls *sort.bam | while read id;do (samtools view -b $id chr17:7661779-7687550 > ${id%%.*}_tp53.bam );done
ls *_tp53.bam |xargs -i samtools index {}
这个 杂合突变 R280T ,需要找到坐标,首先需要知道转录本,有一个网页工具可以继续转换,但是我忘记了。
假装作者是对的,他们的实验的确是引入了这个突变吧。
然后看相关系数
三种文件都准备好了:
首先看 salmon这样的无需比对的流程结果和 hisat2+featureCounts的差异
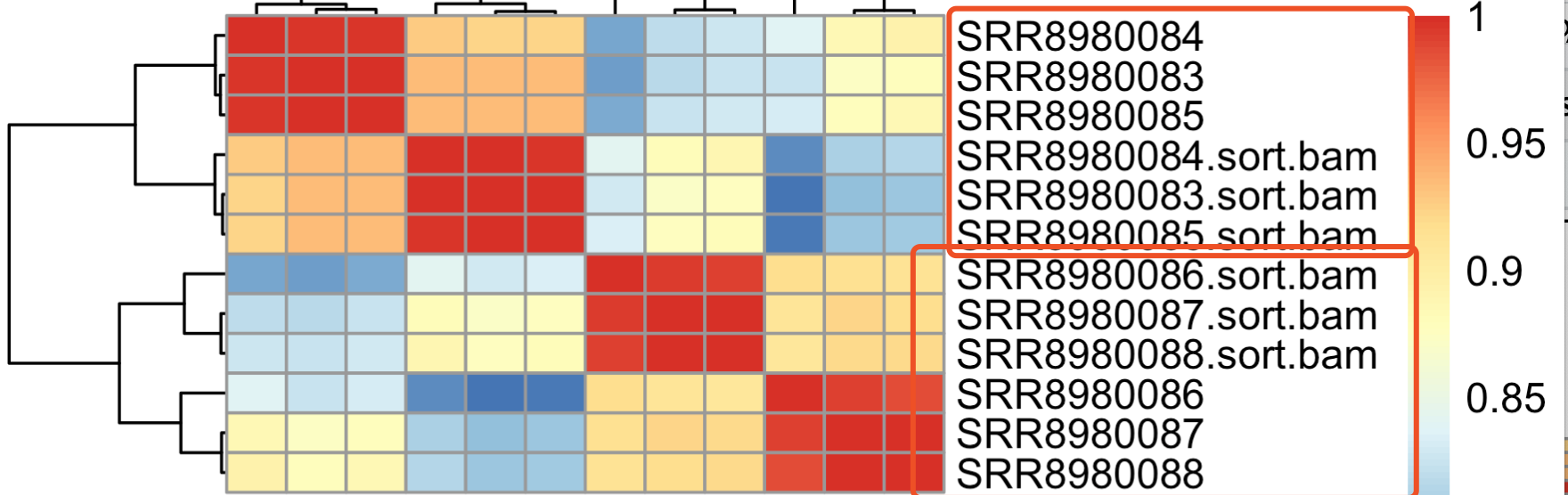
可以看到,同一处理组的样本在不同流程下面得到的表达量直接的相关性,是高于不同组的,符合逻辑!
但是单独查看同一个样本的不同流程的表达量,如下所示:

可以看到,还是有不少基因在不同流程表现差异非常显眼!那同样的,我们需要检查这些基因,简单看看5个差异最大的基因吧。
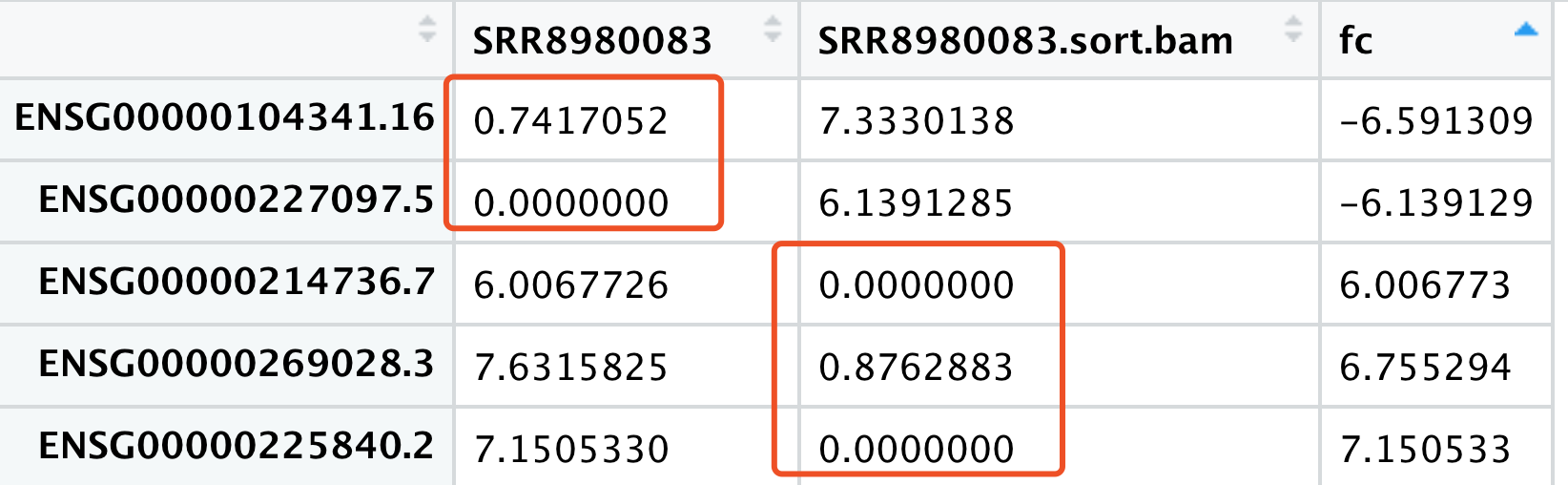
在salmon里面,可以看到第一个基因有3个转录本:
ENST00000445593.6_1 ENSG00000104341.16_2
ENST00000521545.6_1 ENSG00000104341.16_2
ENST00000517924.5_2 ENSG00000104341.16_2
同样的,salmon的这个样本的结果如下:
Name Length EffectiveLength TPM NumReads
ENST00000445593.6 3173 2867.291 0.114792 4.192
ENST00000521545.6 915 609.515 0.000000 0.000
ENST00000517924.5 618 313.557 0.000000 0.000
但是在hisat2+featureCounts的,6个样本都有表达量。
ENSG00000104341.16 chr8;chr8;chr8;chr8;chr8;chr8;chr8;chr8;chr8;chr8;chr8;chr8;chr8;chr8;chr8;chr8;chr8;chr8;chr8;chr8;chr8;chr8;chr8;chr8;chr8;chr8 97775057;97775057;97775776;97775849;97805353;97805353;97805353;97805353;97815304;97815328;97815328;97815328;97816058;97816058;97816058;97816058;97819140;97819140;97819140;97819140;97825058;97825058;97825058;97851397;97851397;97851397 97776108;97776108;97776108;97776108;97805464;97805464;97805464;97805464;97815401;97815401;97815401;97815401;97816180;97816180;97816180;97816180;97819164;97819238;97819238;97819238;97825153;97825153;97825153;97851474;97853013;97852598 +;+;+;+;+;+;+;+;+;+;+;+;+;+;+;+;+;+;+;+;+;+;+;+;+;+ 3197 5453 5150 5346 4250 4775 5271
现在问题是如何判断这个基因是否有表达量,还是需要知道坐标:
chr8 HAVANA gene 97775057 97853013
使用命令去检查bam文件
samtools view SRR8980083.sort.bam chr8:97775057-97853013|wc
发现这个基因的区域的确是有比对成功的reads,这就是我们所说的表达量。
第一个结论,是salmon会漏掉基因的真实表达量。
那么salmon是否会凭空捏造基因的表达量,所以我们还需要检查
chr3 HAVANA transcript 96617188 96618236 . - . gene_id "ENSG00000269028.3"; transcript_id "ENST00000600213.3"; gene_type "protein_coding"; gene_name "MTRNR2L12"; transcript_type "protein_coding"; transcript_name "MTRNR2L12-201"; level 2; protein_id "ENSP00000468991.1"; transcript_support_level "NA"; hgnc_id "HGNC:37169"; tag "not_best_in_genome_evidence"; tag "basic"; tag "appris_principal_1"; havana_gene "OTTHUMG00000175726.3"; havana_transcript "OTTHUMT00000430905.3";
使用命令去检查bam文件
samtools view SRR8980083.sort.bam chr3:96617188-96618236 |wc
的确是没有表达量的,那么为什么salmon会捏造这个表达量呢?