上个月听了jimmy老师的巡讲后,开始学习GEO数据挖掘,看了相关视频后,想实战一把,于是看了jimmy老师去年开始分享的GEO数据挖掘帖子,没想到第一期就碰到个钉子,还好自己解决了,以下是分享。
原帖在GEO数据挖掘-第一期-胶质母细胞瘤(GBM)
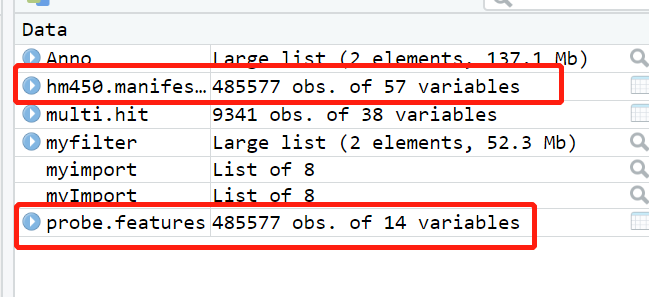
前面都一帆风顺,但是到第二步 得到geneID和gene类型的对应关系时,遇到了钉子
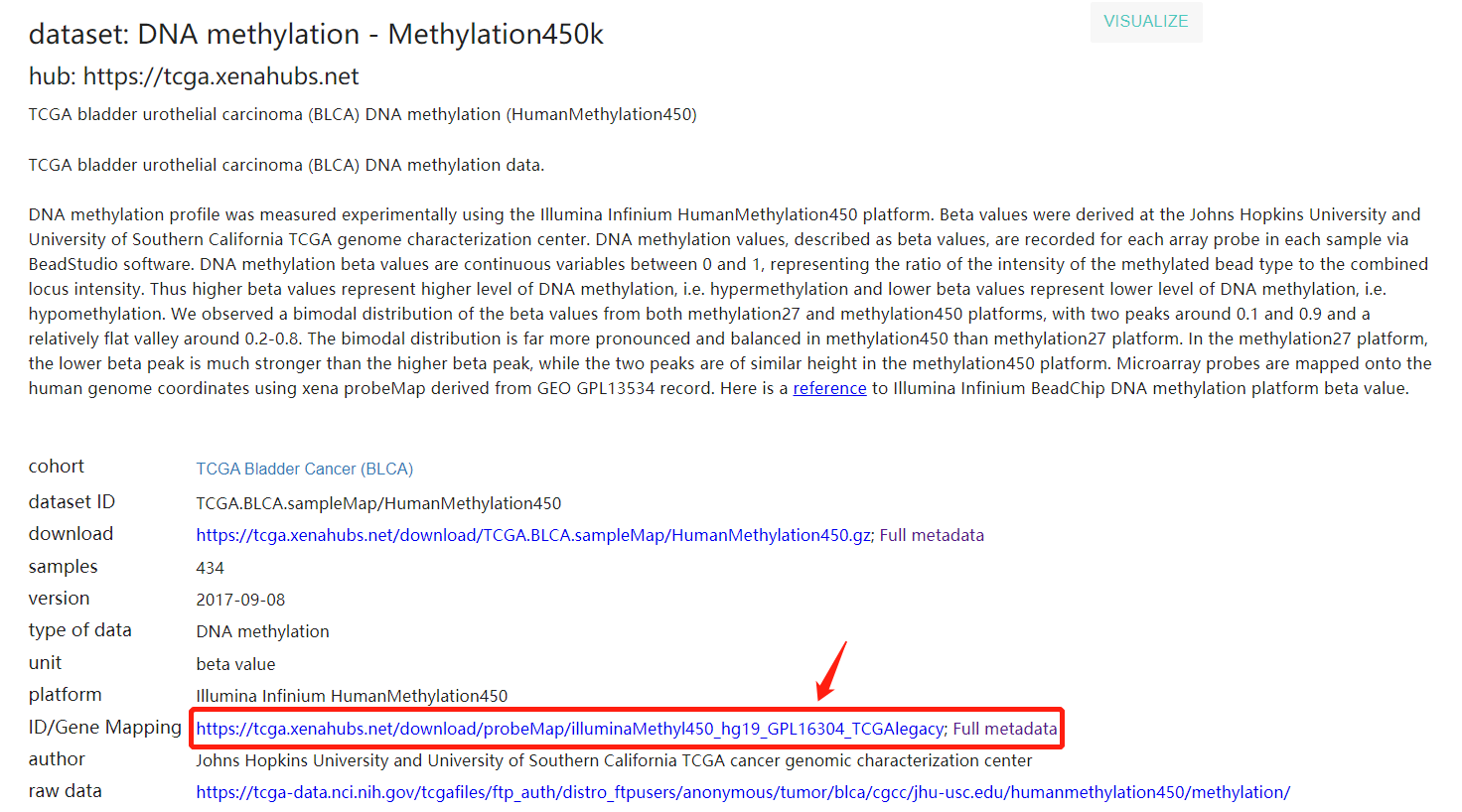
进入帖子说的网站,发现是这样的

于是根据自己的理解,点进去人类的GTF下载界面,发现是这样的
下载了Homo_sapiens.GRCh38.96.gtf.gz这个文件到shell然后解压,如图
好像跟老师原帖里的gtf不太一样,但我的感觉是,既然是这个网站,又是唯一的人类GTF文件,应该没错,于是按原帖的方式转化成了gene2type 格式,读入R,发现不对,于是开始请教jimmy老师,结果。。。

真是个善于诱导(甩锅)学生的好老师。。。
然后,我就开始研究老师原帖给的转化gene2type格式的代码
awk '{if(!NF || /^#/){next}}1'
/public/reference/gtf/gencode/gencode.v25lift37.annotation.gtf | cut -f9 | sed 's/\"//g'| sed 's/;//g' | awk '{ print $4"\t"$8 }' |awk '{if(/^E/){next}}1'| awk '{ print $2"\t"$1 }' | sort -k 1 | uniq > gencode.v25lift37.annotation.gtf.gene2type
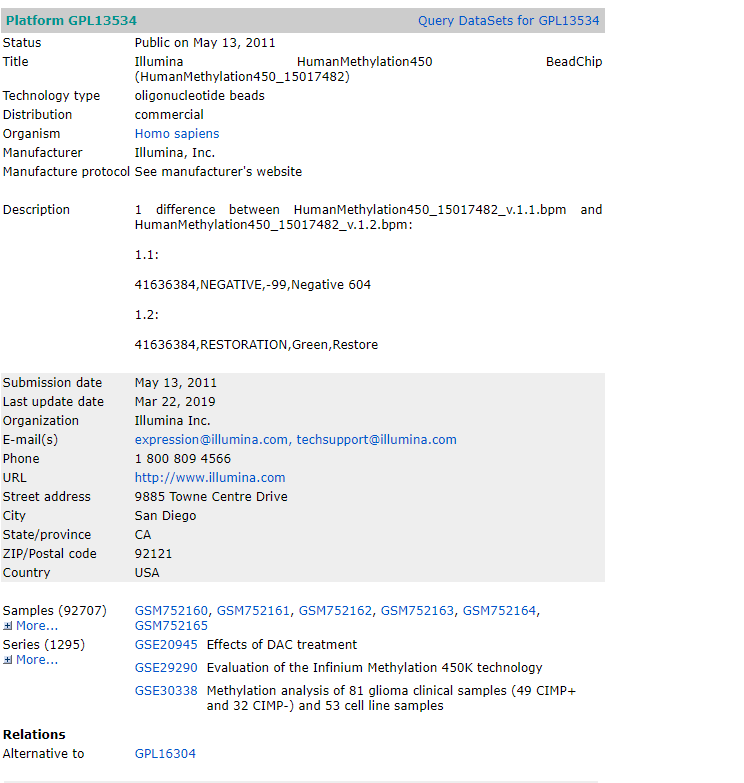
有所发现,需要的gtf其实就是gencode.v25lift37.annotation.gtf 这个文件,于是google了一哈,发现应该是在genecode 这个网站
于是点了右侧的GTF 按钮下载,然后按照源代码,修改转化文件的参数,转化为gene2type
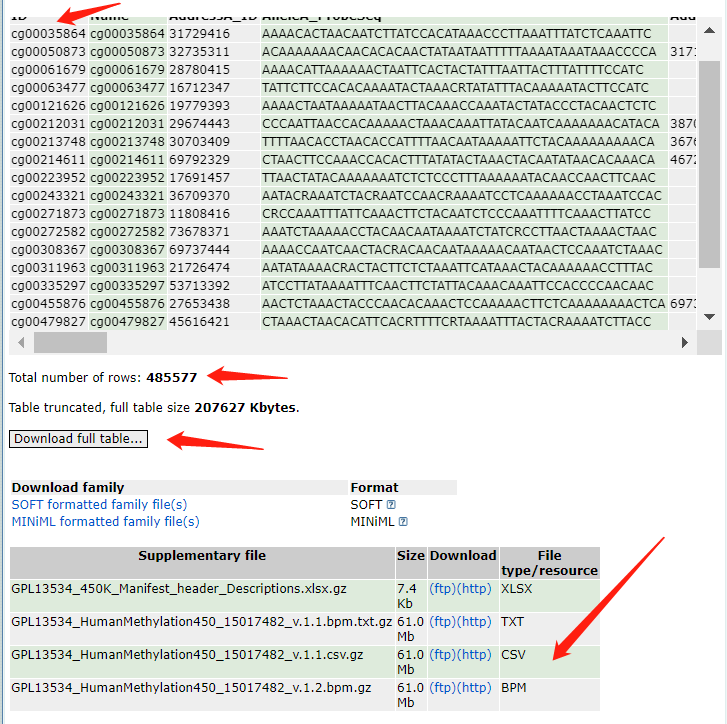
这还没结束,导入R,发现有点不对劲
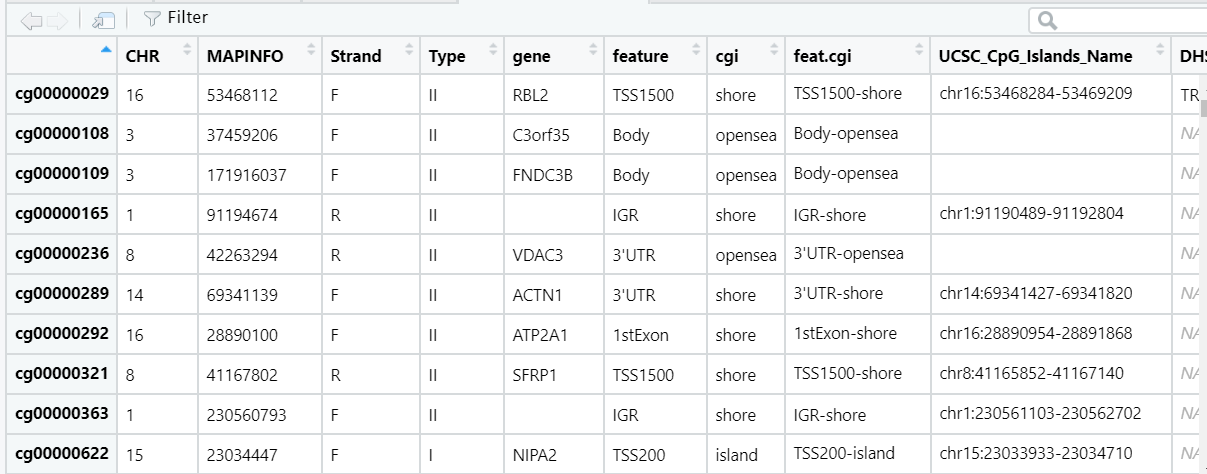
可以看到数目偏少(只有7641个),且gene和type出现了很多奇怪的内容,感觉肯定是转化的时候出问题了
然后又仔细研究了一下下载的网站,发现可以找到老师原帖的历史版本gtf文件
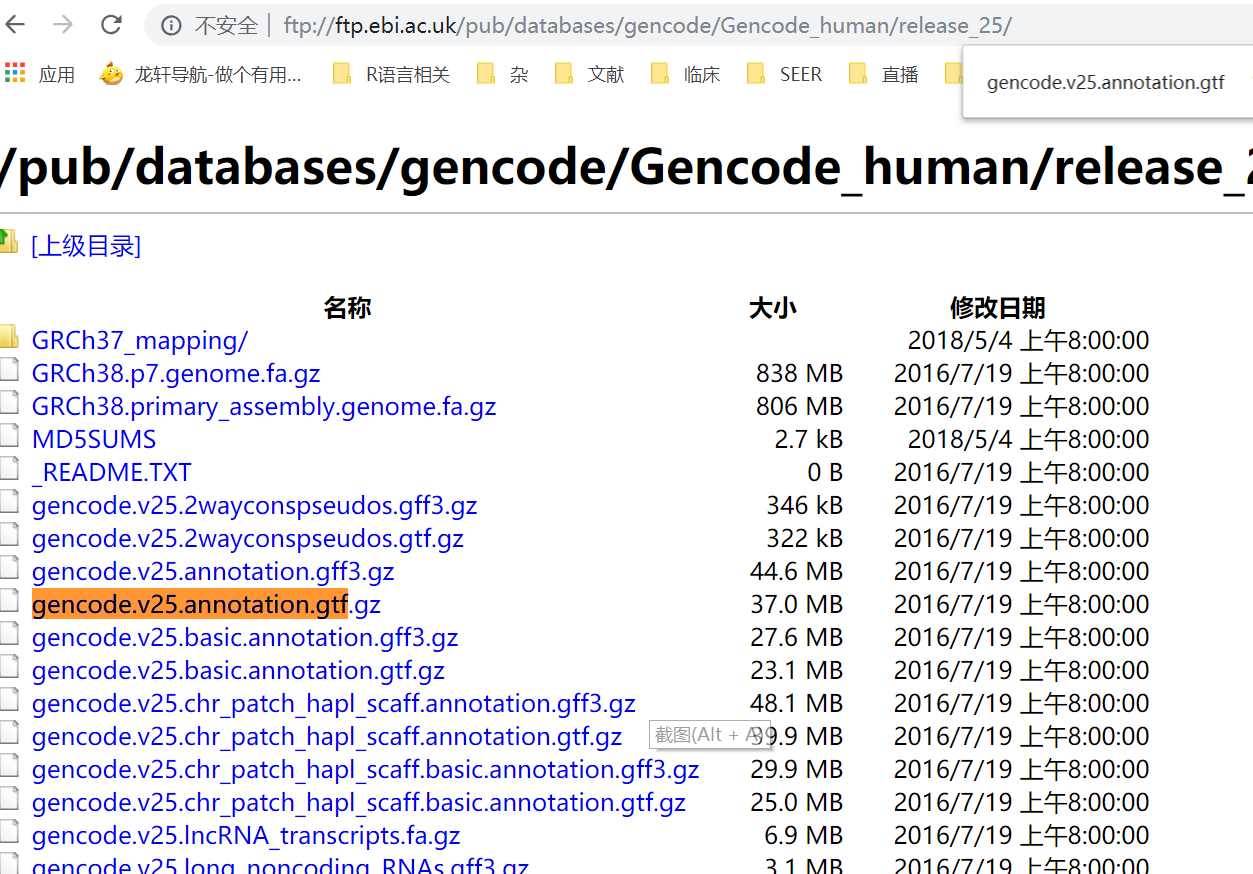
于是下载下来,跟新版的gtf文件对比,cat一下
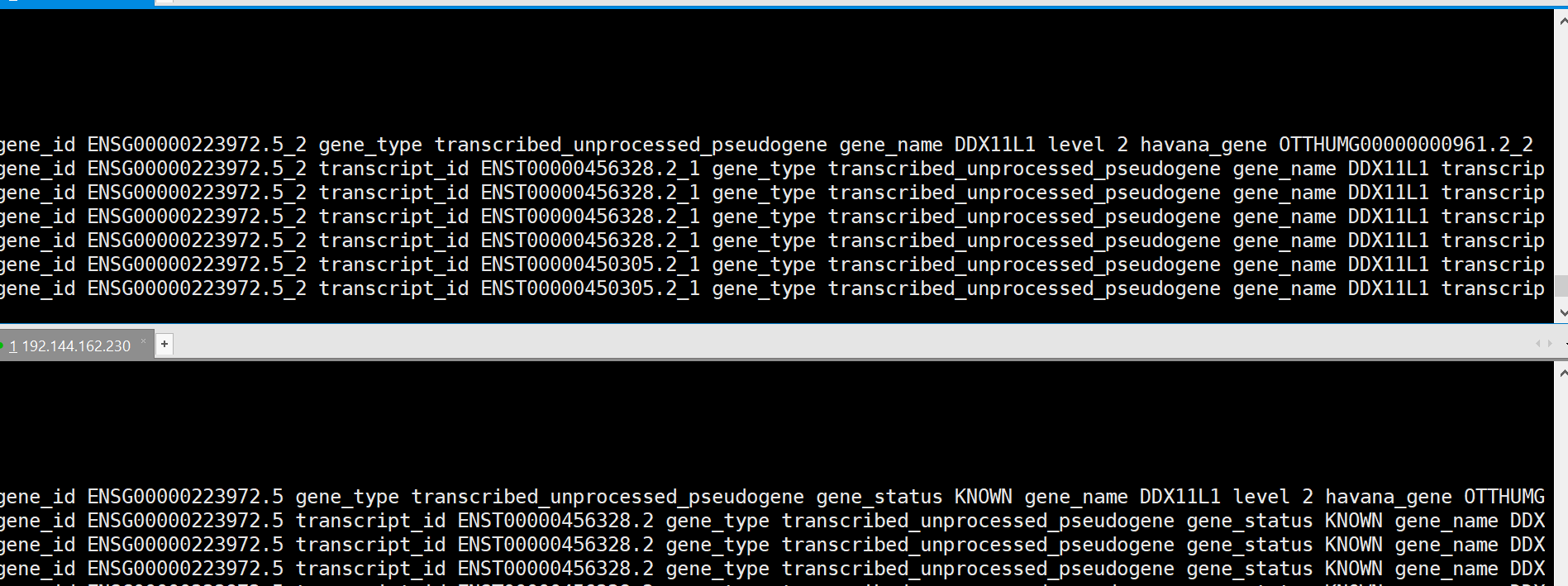
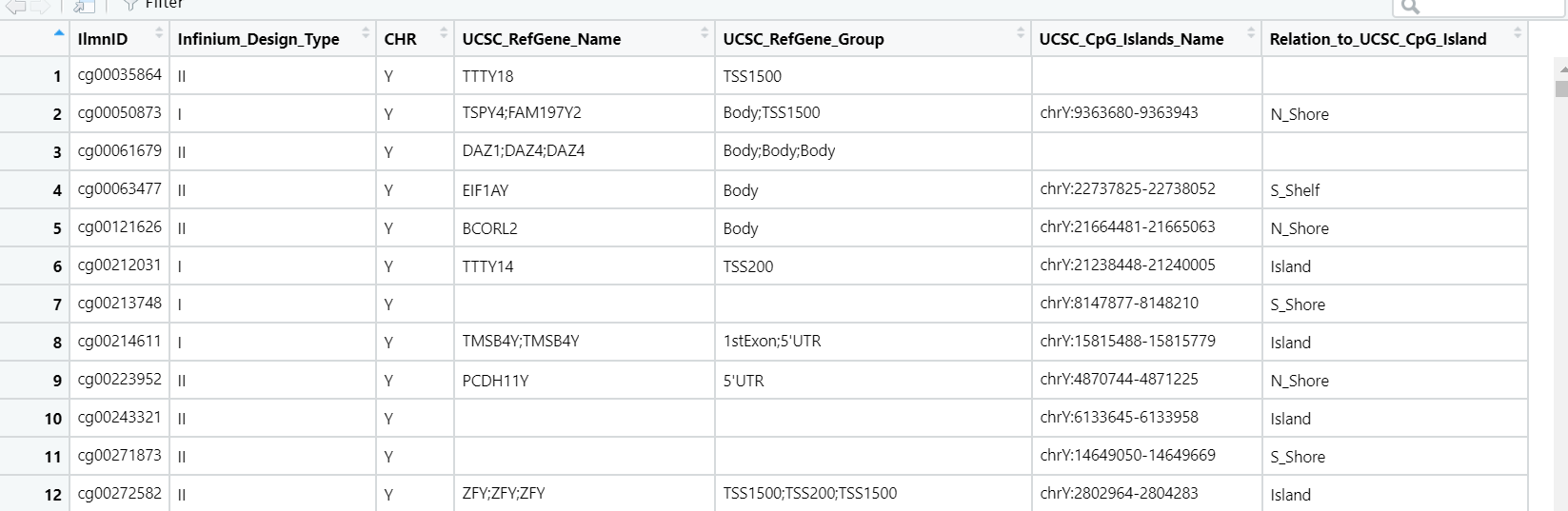
发现,上面的是最新版的gtf,下面是老师写教程用的gtf,两者对比一下之后发现,只是最新版的gtf没有gene_status这一个项目,其他都一样,于是就简单了,修改下代码取得相应列数,提取gene_type和gene_name 就ok咯
awk '{if(!NF || /^#/){next}}1' gencode.v30lift37.annotation.gtf | cut -f9 | sed 's/\"//g'| sed 's/;//g' | awk '{ print $4"\t"$6 }'|awk '{if(/^E/){next}}1'| awk '{ print $2"\t"$1 }' |sort -k 1 | uniq > gencode.v30new.annotation.gtf.gene2type
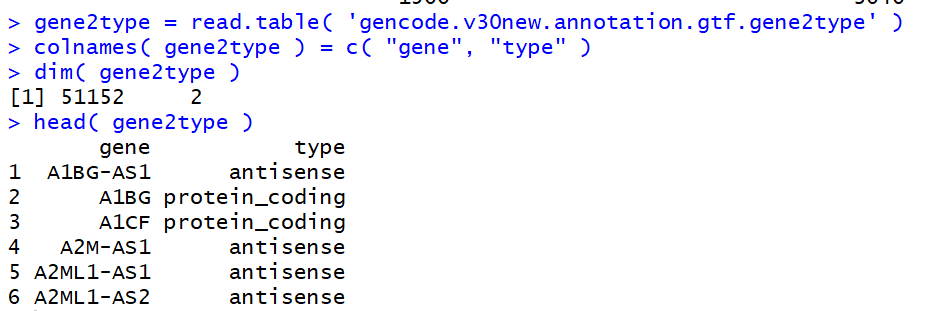
再导入R,一看,大功告成!
真是万事开头难,然后,残忍的jimmy老师竟然要我把这段探索经历 投稿给他做教程,我晕,还得自学下markdown ,我这么小的事,发出来肯定没人看啊,但是,看了下原帖的留言,我改变了看法


问这个开头第一步的同学还不只我一个。。。看来我这个探索经历还是挺有意义的哈哈
心得
最后是自己的一点体会吧,完事开头难,学生信需要智慧,更需要毅力和不断地探索,一个小的不能再小的问题也值得自己好好琢磨,你看看我,被jimmy老师一逼,起码,一,搞懂了两个网站以及GTF数据的下载和内容概要读取;二,转化文件的时候重温了下shell语言;最后,竟然还顺便学会了用markdown 交作业!!哈哈哈