看过单细胞水平的癌症研究的朋友都应该是对下面这幅图不陌生:
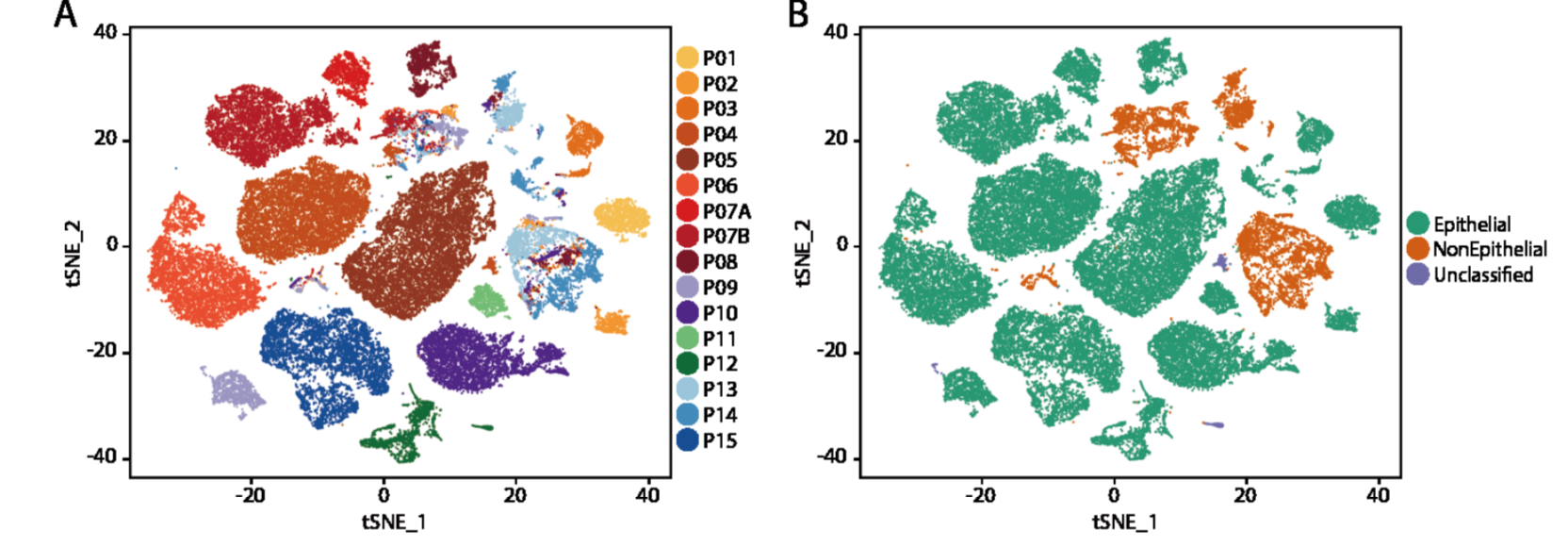
可以很清晰的看到,多个病人的单细胞可以分成恶性的上皮细胞和非恶性的肿瘤微环境,微环境的那些细胞可以聚集成为很多类,而且每个类别的细胞都是来源于不同病人的。
但是呢,对于那些肿瘤恶性细胞,也是聚集为很多类别,但基本上每个类别都是一个单独的病人来源。
单细胞转录组数据分析CNV判断恶性与否
单细胞转录组数据分析CNV最早是aviv Regev实验室提出,他们的一系列CNS文章都利用了单细胞转录组数据分析CNV,我在单细胞天地发布了一系列教程。
- 单细胞转录组数据分析CNV
- 使用broad出品的inferCNV来对单细胞转录组数据推断CNV信息
- 使用inferCNV来推断CCLE转录组数据的拷贝数变异
- 使用inferCNV来推断2014的science关于GBM文章的单细胞转录组数据的拷贝数情况
我也拿那个软件在普通的bulk转录组数据,CCLE数据库数据,以及两个单细胞数据集测试了,最后在2014的science关于GBM文章的数据里面验证了。
我在单细胞天地解读的30多篇这样的研究,都看到了这个现象,本来是默认一个规律,我心里倒是起过涟漪,应该是需要探索一下,开启一个课题。
看到一个很有趣的研究,研究者的出发点,并不是要探索为什么聚类的时候肿瘤细胞之所以在不同病人完全分开,而非肿瘤细胞不会有病人特异性,但是却从代谢角度无意中回答了这个问题!两个数据集
单细胞领军人物aviv Regev自己实验室发布了一系列肿瘤单细胞研究,包括:
- 首先是2014年关于GBM的science文章;PMID: 24925914 ,提到了这个单细胞转录组数据分析CNV
- 2016年关于melanoma的science文章:PMID: 27124452 也应用了单细胞转录组数据分析CNV,该文章的数据公布在 GSE72056 这次使用的Smart-seq2建库技术,共计 4645 个细胞,仅仅是表达矩阵就有71Mb
- 2016年CELL杂志发表的关于头颈癌的文章:Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer 测序数据是:We profiled transcriptomes of ∼6,000 single cells from 18 head and neck squamous cell carcinoma (HNSCC) patients, including five matched pairs of primary tumors and lymph node metastases.
而这篇文章使用的是2016的melanoma和HNSCC数据,在GEO上面是: GSE72056and GSE103322.
拿到表达矩阵和原作者非常完善的注释信息后,就可以很容易看看恶性的肿瘤细胞和肿瘤微环境细胞根据代谢相关基因的分类结果。( 总共是1566个基因,来自于 85 metabolic pathways (from KEGG) )

后面的分析更精彩
主要是探索不同细胞类型、细胞亚型的代谢特点和代谢通路的异质性,揭示了肿瘤代谢异质性在单细胞水平上表现出的与组织水平上的显著区别,表明单细胞水平上的研究对于深入理解肿瘤微环境的代谢异质性至关重要。 但却不是我感兴趣的。
最后,留一个彩蛋,这个背后还有一个算法故事,就是抹去了肿瘤病人特异性后,剩下的肿瘤细胞的表达信息是可以把肿瘤进行新的分组,那些分组会优于一切之前的研究,最起码是CNS文章,你要不要试试看?