以前下载TCGA数据,喜欢使用UCSC的XENA数据库, 全部数据在:https://xenabrowser.net/datapages/ 这个时候有两个数据源,需要区分开来;
- GDC TCGA Breast Cancer (BRCA)&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443) (18 datasets) 09-15-2017
- TCGA Breast Cancer (BRCA)&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443) (30 datasets) 2016-04-27
前者数据是 IlluminaHiSeq TCGA hub表达矩阵,基因SYMBOL的表达矩阵,基因的表达信息,通常是用来把病人进行分组,之后还需要下载临床信息,才能做生存分析。
分分钟对TCGA数据库的任意癌症种类做生存分析,并校验
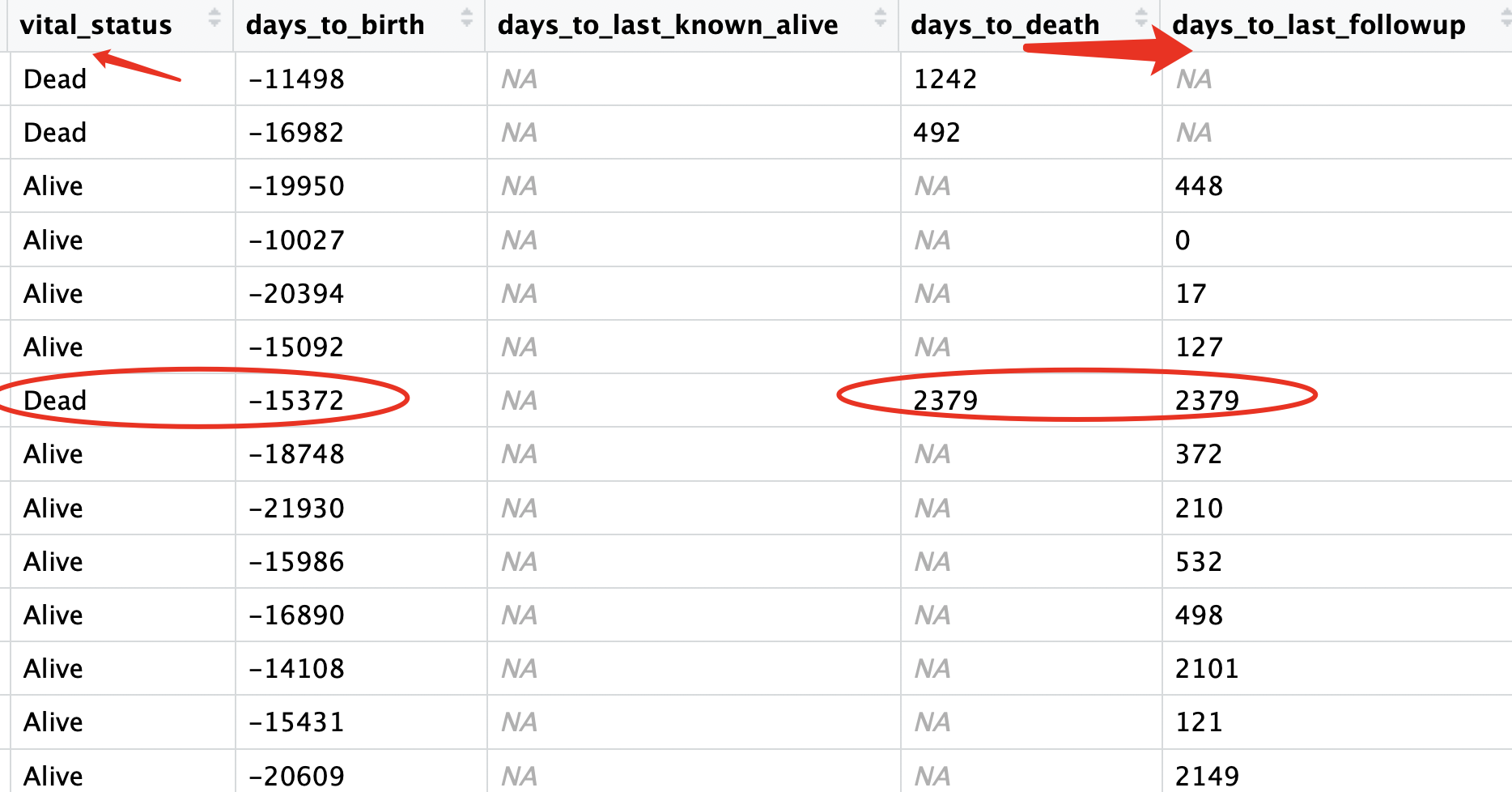
发现TCGA数据库记录病人的生存事件的时候,区分Alive和Dead,但是呢,不同的事件本来是应该对应不同的时间记录字段,但是突然就发现了一个特例,虽然不清楚为什么,但是毫无疑问我们的代码需要注意这一点了。
推测可能是因为一个病人有多个样本,不同样本记混了。
现在我喜欢使用TCGAmutations下载突变数据和临床信息,代码如下:
library(TCGAmutations)
tcga_available() #查看可用的数据
tcga_load(study = "LGG") # 默认加载经过校正后的MC3 maf文件
tcga_mc3=tcga_lgg_mc3
laml <- tcga_mc3
phe=as.data.frame(laml@clinical.data)
初步下载得到的phe,就是上面那样的不合理数据,需要进行校正,更有趣的是这个信息其实要比XENA来说,过时一点,大家可以自行去比较。
构建生存分析需要的时间
我这里使用的代码好像很复杂:
table(phe$vital_status)
phe=phe[phe$vital_status %in% c('Alive' , 'Dead'),]
phe$time=0
pos=which(phe$vital_status=='Dead')
phe$time[pos]=phe$days_to_death[pos]
pos=which(phe$vital_status=='Alive')
phe$time[pos]=phe$days_to_last_followup[pos]
phe=phe[,c("Tumor_Sample_Barcode","gender","vital_status",'time')]
phe$vital_status=ifelse(phe$vital_status=='Alive',0,1)
最后得到的phe如下所示:
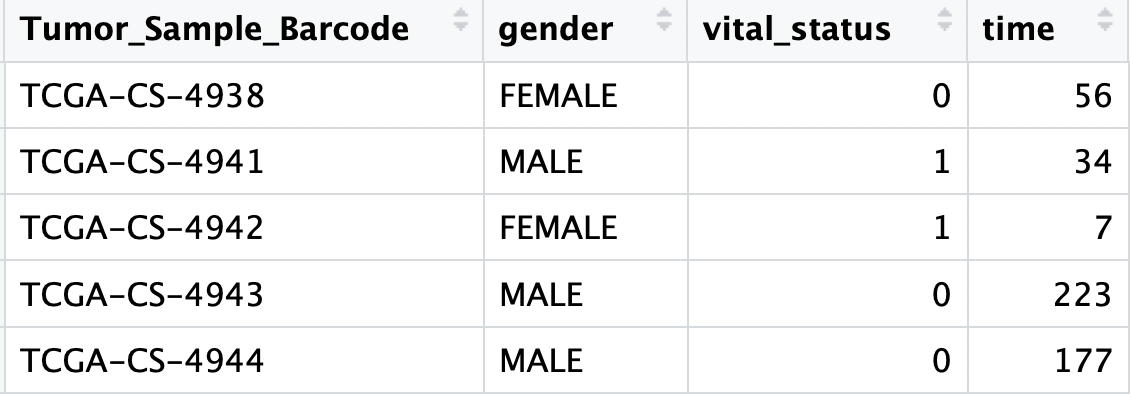
需要留意的是,同一个病人因为有多个测序样品,如果不是12位的ID,比如15位的ID,就需要去冗余:
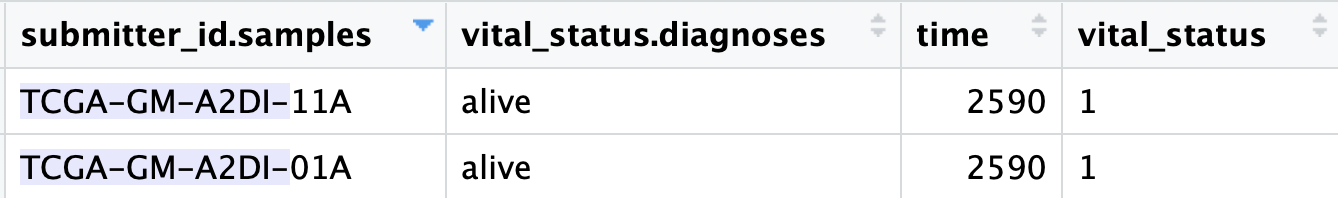
上面的例子就是一个病人被记录了两次临床信息,仅仅是因为他有两个转录组测序而已,这个时候一个病人的两个样本都被记录临床信息,好的情况是记录的都是一致的,因为大家共享一个病人ID。但是也有情况出现就是他们不一致,所以就出现了bugs
生存分析代码是
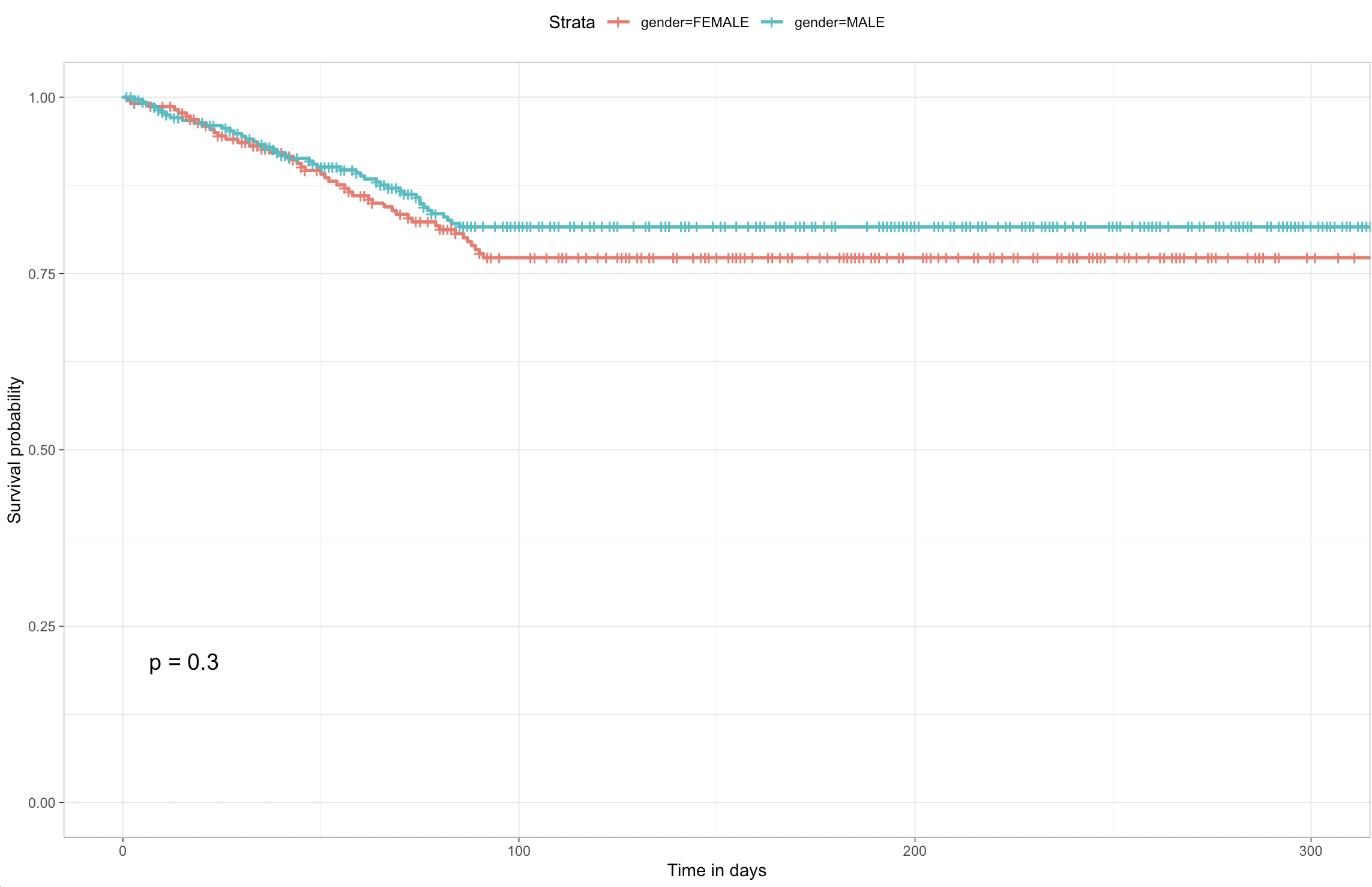
有了上面的数据, 就可以做生存分析并且绘制代码了。
library(survival)
library(survminer)
mySurv_OS=with(phe,Surv(time, vital_status))
sfit=survfit(mySurv_OS~gender,data=phe)
p=ggsurvplot(sfit,data = phe,
pval =TRUE,
xlab ="Time in days",
ggtheme =theme_light())
print(p)
可以看到,性别的生存差异不显著: