大家都喜欢整合多组学数据,实际上目前大多数研究都是拿其中一种组学数据来对样本进行分类,然后查看病人分组后另外一种组学的差异情况。再其次,就是整合多组学数据对样本统一分组。
案例介绍
看到文章 Integrative analysis of the inter-tumoral heterogeneity of triple-negative breast cancer 针对137个TNBC病人的3种数据,进行挑选后,各自进入NMF聚类:

发现3种数据重合度很少 ,最后NMF-gene (1187 genes), NMF-miRNA (61 miRNAs), and NMF-CNV (2044 CNVs)可视化如下:

所以使用了SNFtool包整合多组学数据进行病人分组,如下:
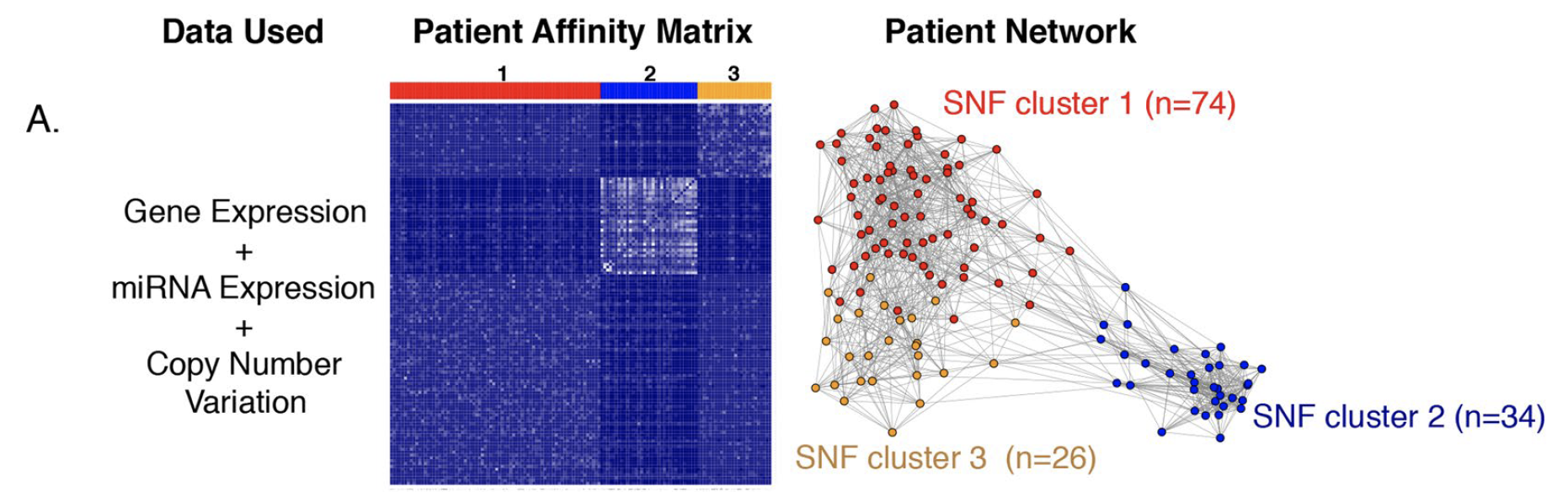
既然是区分成为3组,那就必须使用公共数据库来说明这样的分组是有显著的生存意义的。
下面我们看看这个包的用法。
包的使用帮助
有趣的是,这个包并不是在bioconductor,而是在cran上面。https://cran.r-project.org/web/packages/SNFtool/index.html
SNF tool is an R package for Similarity Network Fusion: taking multiple views of a network and merging them into a combined view.
This repo is a fork from https://github.com/cran/SNFtool with added functionality and documentation. More information on the original version can be found at http://compbio.cs.toronto.edu/SNF/SNF/Software.html
似乎也没有看到这个包发文章,但是看到一些文章引用它,比如:BMC Med Genomics. 2016; doi: 10.1186/s12920-016-0192-7深度学习整合多组学数据
发表于 March 2018,题目是:Deep Learning–Based Multi-Omics Integration Robustly Predicts Survival in Liver Cancer 使用的就是TCGA HCC cohort (360个病人), 这里选取了3种数据,mRNA, DNA methylation and miRNA 首先走deep Learning流程 (就是简单的keras)
值得一提的是,这里的methylation数据,是把基因的TSS前面1.5kb的探针取平均值后算作是基因的甲基化水平。
本文的deep Learning流程输入数据的 15,629 genes from RNA-seq, 365 miRNAs from miRNAseq, and 19,883 genes from DNA methylation data
走完deep Learning流程,最后可以得到 two survival risk subtypes同样的分析策略很容易应用到其它癌症
比如发表于 Front Genet. 2018; Deep Learning-Based Multi-Omics Data Integration Reveals Two Prognostic Subtypes in High-Risk Neuroblastoma
