在教师节收到学生的提问,刷我B站74小时视频的时候看到我演示了RNA-seq差异分析只用了一行代码就完成了3大R包的全部分析,并且输出了对应的图表结果,觉得很神奇,但是B站视频并没有配套讲义和代码还有测试数据。
首先我一直使用airway数据集做测试
airway数据集在这里我就不多说了,搜索生信技能树早期教程可以看到很多介绍。
## 表达矩阵来自于R包: airway
if(F){
library(airway)
data(airway)
exprSet=assay(airway)
group_list=colData(airway)[,3]
save(exprSet,group_list,file = 'airway_exprSet.Rdata')
}
load(file = 'airway_exprSet.Rdata')
if(T){
colnames(exprSet)
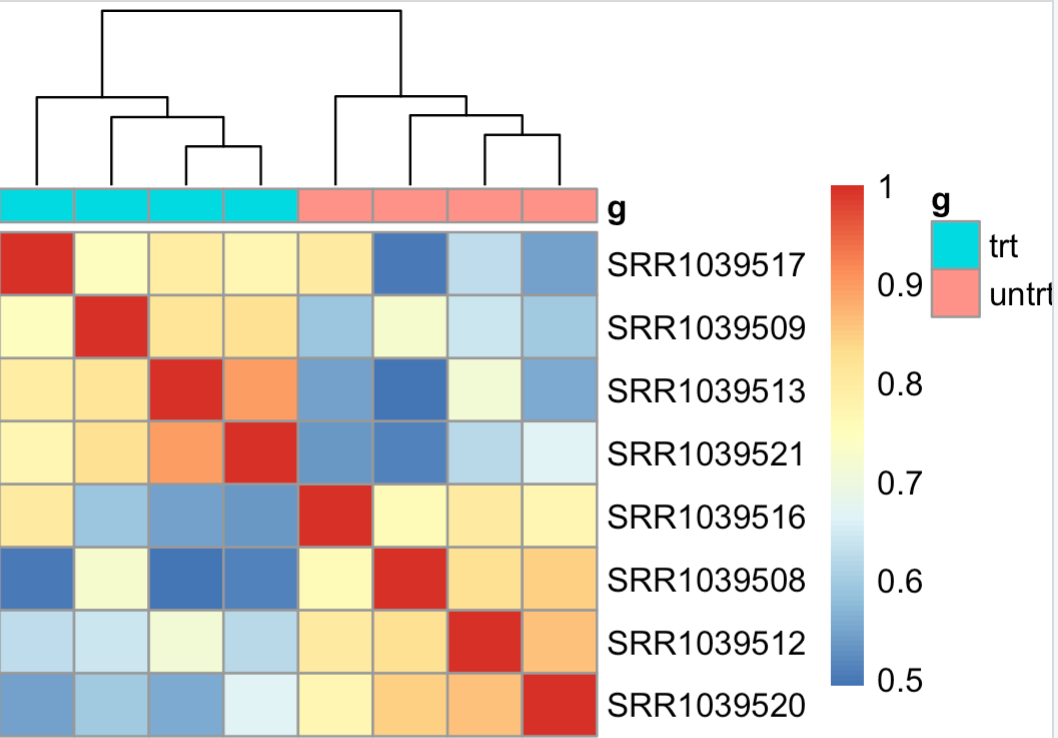
pheatmap::pheatmap(cor(exprSet))
group_list
tmp=data.frame(g=group_list)
rownames(tmp)=colnames(exprSet)
# 组内的样本的相似性应该是要高于组间的!
pheatmap::pheatmap(cor(exprSet),annotation_col = tmp)
dim(exprSet)
exprSet=exprSet[apply(exprSet,1, function(x) sum(x>1) > 5),]
dim(exprSet)
exprSet=log(edgeR::cpm(exprSet)+1)
dim(exprSet)
exprSet=exprSet[names(sort(apply(exprSet, 1,mad),decreasing = T)[1:500]),]
dim(exprSet)
M=cor(log2(exprSet+1))
tmp=data.frame(g=group_list)
rownames(tmp)=colnames(M)
pheatmap::pheatmap(M,annotation_col = tmp)
pheatmap::pheatmap(M,annotation_col = tmp,filename = 'cor.png')
library(pheatmap)
pheatmap(scale(cor(log2(exprSet+1))))
}
很明显可以看到, 组内的样本的相似性应该是要高于组间的!
而且为了显示这个规律,我还做了一个统计学技巧展示,当然了,很多人非常的不用心,所以把视频听10遍也看不懂,get不到我的点,需要批评!
使用我包装好的函数即可
可以看到,下面的代码非常简洁,因为仅仅是使用了 run_DEG_RNAseq 函数,就根据表达矩阵和分组信息,完成了全部的分析!
rm(list = ls())
options(stringsAsFactors = F)
load(file = 'airway_exprSet.Rdata')
group_list
group_list=relevel(group_list,ref = 'untrt')
source('run_DEG_RNA-seq.R')
run_DEG_RNAseq(exprSet,group_list,
g1="untrt",g2="trt",
pro='airway')
这就是大家看视频后提的问题,为什么这么神奇呢?下面的图表是如何自动出来的呢?

因为这个 run_DEG_RNAseq 函数的代码非常长,这里我就不贴在公众号了哈,大家可以在我的GitHub的GEO项目找到它!
GEO传奇代码
一不留神,这个GEO项目就成为了点赞数最多的,直接孵化出12篇数据挖掘类SCI文章,至于间接的那些就不计其数了,因为大家都是偷偷的使用,也不告诉我,甚至某些别有用心者还不告诉身边的人,要一个人独享这些代码。
https://github.com/jmzeng1314/GEO
https://github.com/jmzeng1314/GEO/tree/master/airway_RNAseq
既然是多个R包,结果该如何取舍呢?
这个时候是没有标准答案的,因为每个R包都非常热门,引用量都是好几千,你选择哪个都符合市场规律,不过,我这里有一个代码,对3个结果根据阈值筛选交集。
https://github.com/jmzeng1314/GEO/tree/master/airway_RNAseq
差异基因后是不是也可以批量GO/KEGG数据库注释呢?
当然是啊,都会写代码了,还有什么是不能为所欲为的呢?
同样的,代码也是在GitHub,需要你仔细理解,不过我有一个小小的要求,请不要把我的代码雪藏,或者刻意隐瞒。
https://github.com/jmzeng1314/GEO/tree/master/airway_RNAseq
值得一提的是这里面的一行代码是需要格外注意的哦:
group_list=relevel(group_list,ref = 'untrt')