今天单细胞授课现场差点翻车,最后做完几个基因集的批量超几何分布检验,想现场解释一波这个富集分析结果的一些数字,如下:

23239是小鼠背景基因,可以理解为物种的基因总数,然后1240是我们感兴趣的基因的总数,可以理解为差异表达分析得到的基因数量,所以对每个通路都是一样的,这个时候我想解释一波,每个GO基因集的数量是如何来的。
拿到指定功能基因集的数量
这里简单使用R包org.Mm.eg.db来获取,代码如下:
library(GO.db)
ls("package:GO.db")
library(org.Mm.eg.db)
go2gene=toTable(org.Mm.egGO2ALLEGS)
this_go_this_gene=go2gene[go2gene$go_id=='GO:0140014',]
table(this_go_this_gene$Evidence)
得到的数据框是:
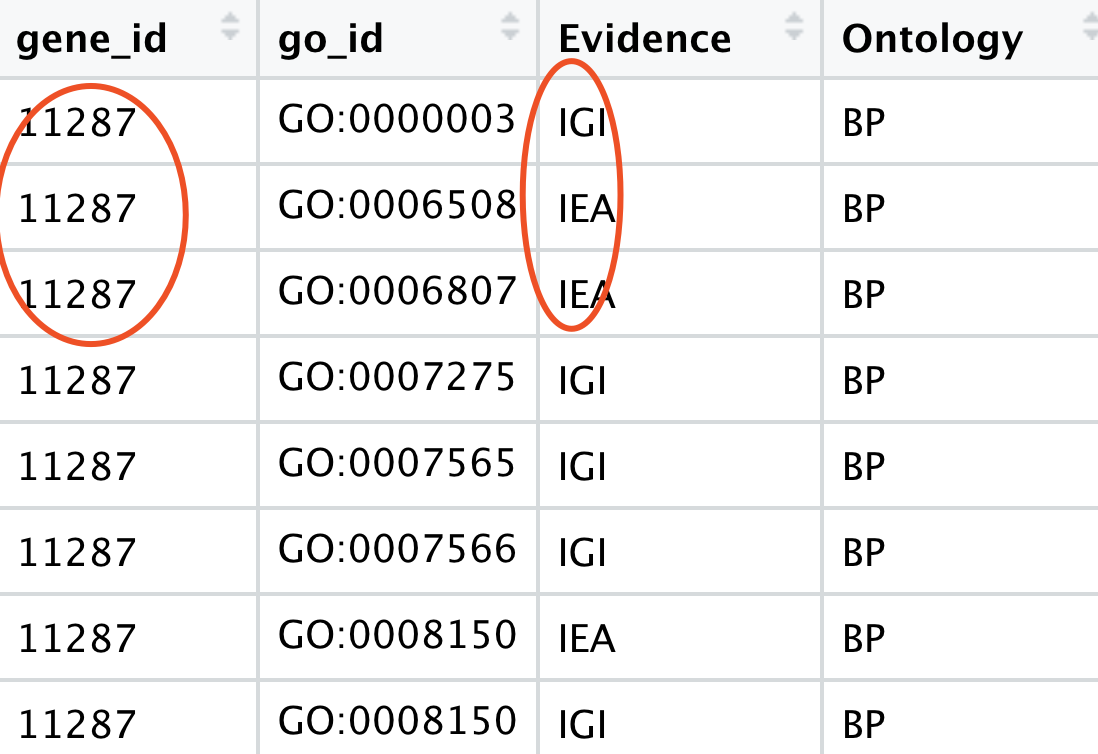
这样得到了
library(GO.db)
ls("package:GO.db")
library(org.Mm.eg.db)
go2gene=toTable(org.Mm.egGO2ALLEGS)
this_go_this_gene=go2gene[go2gene$go_id=='GO:0140014',]
table(this_go_this_gene$Evidence)
这样得到了GO:0140014的全部基因,跟大家去谷歌搜索GO:0140014效果一样,但是呢,看了看是348列,并不是272,这个时候我做了一个错误的判断:我认为是evidence需要筛选。
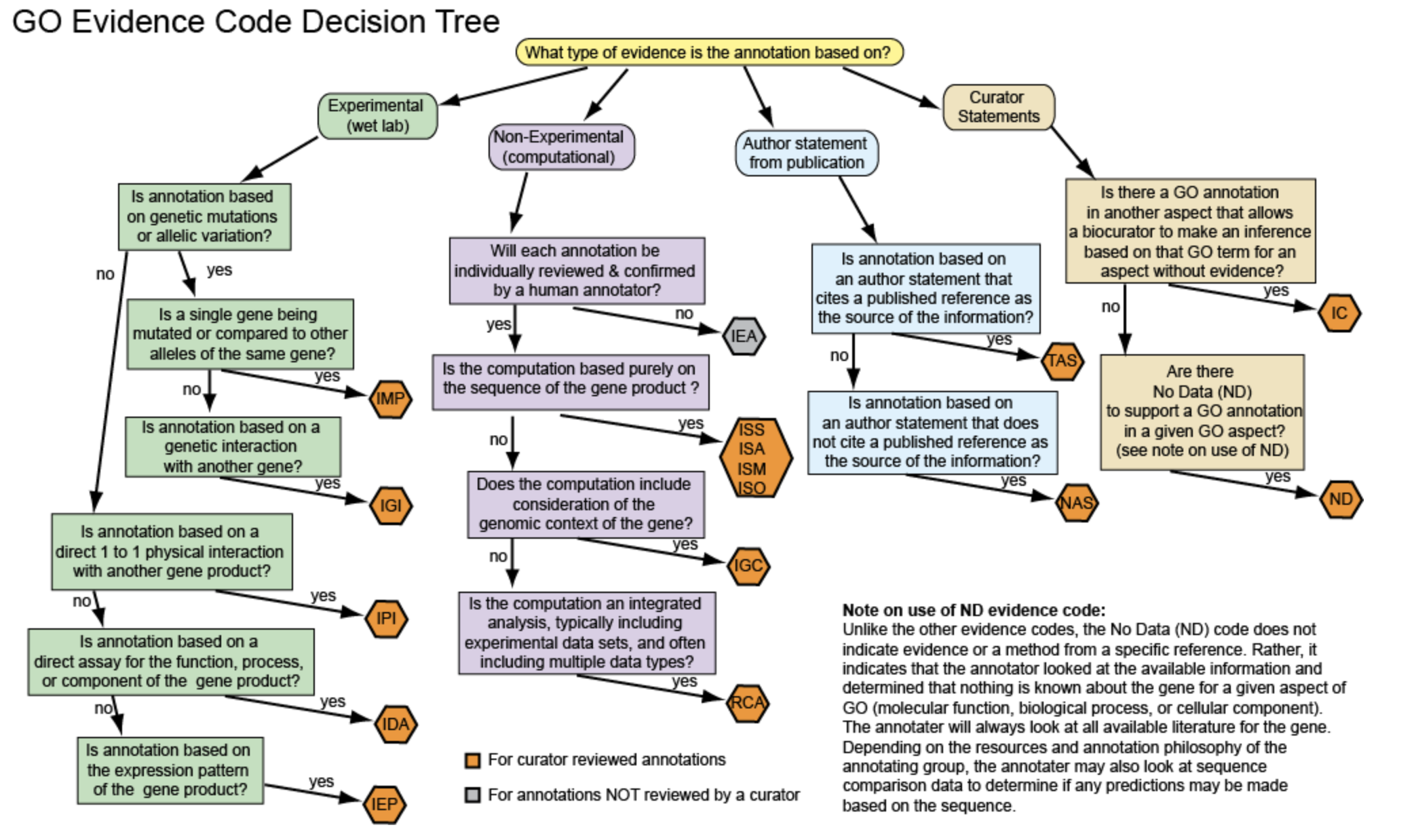
不同的证据支持区别是?
浏览wiki可以看到,是非常的复杂,如下;

时间关系,来不及具体看中文介绍,就打马虎眼略过了,不然单细胞课程就没得上了,仅仅是讲解GO数据框就可以讲一整天。
马前失蹄在于?
实际上,我关注了变化的那一列,就是evidence,却忽略了没有变的那个列,就是基因ID,也就是说一个基因在这个数据框出现多次,我不应该数数据框的行,而是数基因的去冗余后个数。
这样就是正确数值了。
留一个悬念
小鼠这个物种的背景基因数量是23239个,是如何计算的呢,基于什么数据框呢?