每月一生信流程栏目灵感来自于《铁汉1991》博客的《每日一生信》,他那个时候介绍的主要是生信基础知识,包括数据结构,数据格式,数据库资源,计算机基础等等,所以每天都可以进步,每天都有成果。这些基础知识已经被分享的七七八八了,所以我这里推陈出新,来一个每月一生信流程,陪生信技能树的粉丝们一起进步!
上一期我们推荐的是转录组经典表达量矩阵下游分析大全 本期我们聊聊可变剪切,流程里面写的差异转录本,或者差异外显子,都差不多的意思。全部bioconductor流程链接在;http://www.bioconductor.org/packages/release/BiocViews.html#___GeneExpressionWorkflow 目前的27个流程,已经分门别类的整理好了,我们每个月学一个流程,预计两年就可以成为生物信息学领域的全栈工程师啦!
一图看懂DGE, DTE and DTU
参考文献:F1000Research 2019, 8:213 Last updated: 18 MAR 2019
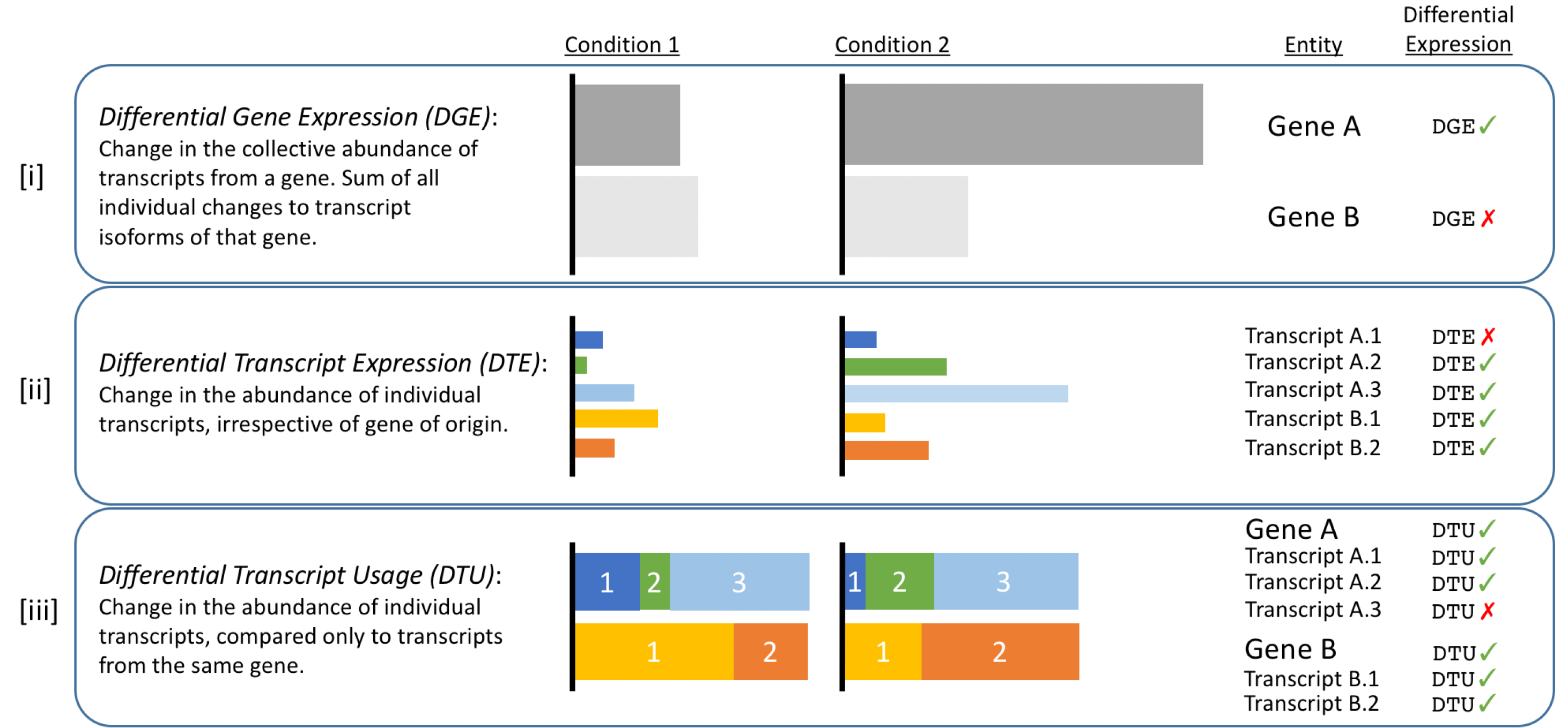
今天学习rnaseqDTU
我们首先看看转录组领域的基因表达相关流程吧,首先一起学习的是Swimming downstream: statistical analysis of differential transcript usage following Salmon quantification
这个就没有中文版了,实际上是使用salmon软件把我们的RNA-seq测序的fastq数据根据参考转录组进行定量后,走几个R包流程。
在R里面安装这个bioconductor流程
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("rnaseqDTU")
因为原文写的实在是太详细了,我这里就不拷贝粘贴了,大家直接去阅读即可:
- RNA-seq workflow for differential transcript usage following Salmon quantification
- http://www.bioconductor.org/packages/release/workflows/vignettes/rnaseqDTU/inst/doc/rnaseqDTU.R
全部目录如下;
- 3 Methods
- 4 Workflow
- 5 Statistical analysis of differential transcript usage
-
6 DTU analysis complements DGE analysis
- 2 背景介绍
- 3 初始配置
- 4 数据整合
- 5 数据预处理
- 6 差异表达分析
- 7 使用camera的基因集检验
流程代码
首先是salmon的:
salmon quant -i $index -l A -1 $fq1 -2 $fq2 -p 4 -o quants/${sample}_quant
每个样本的fq测序数据都会被
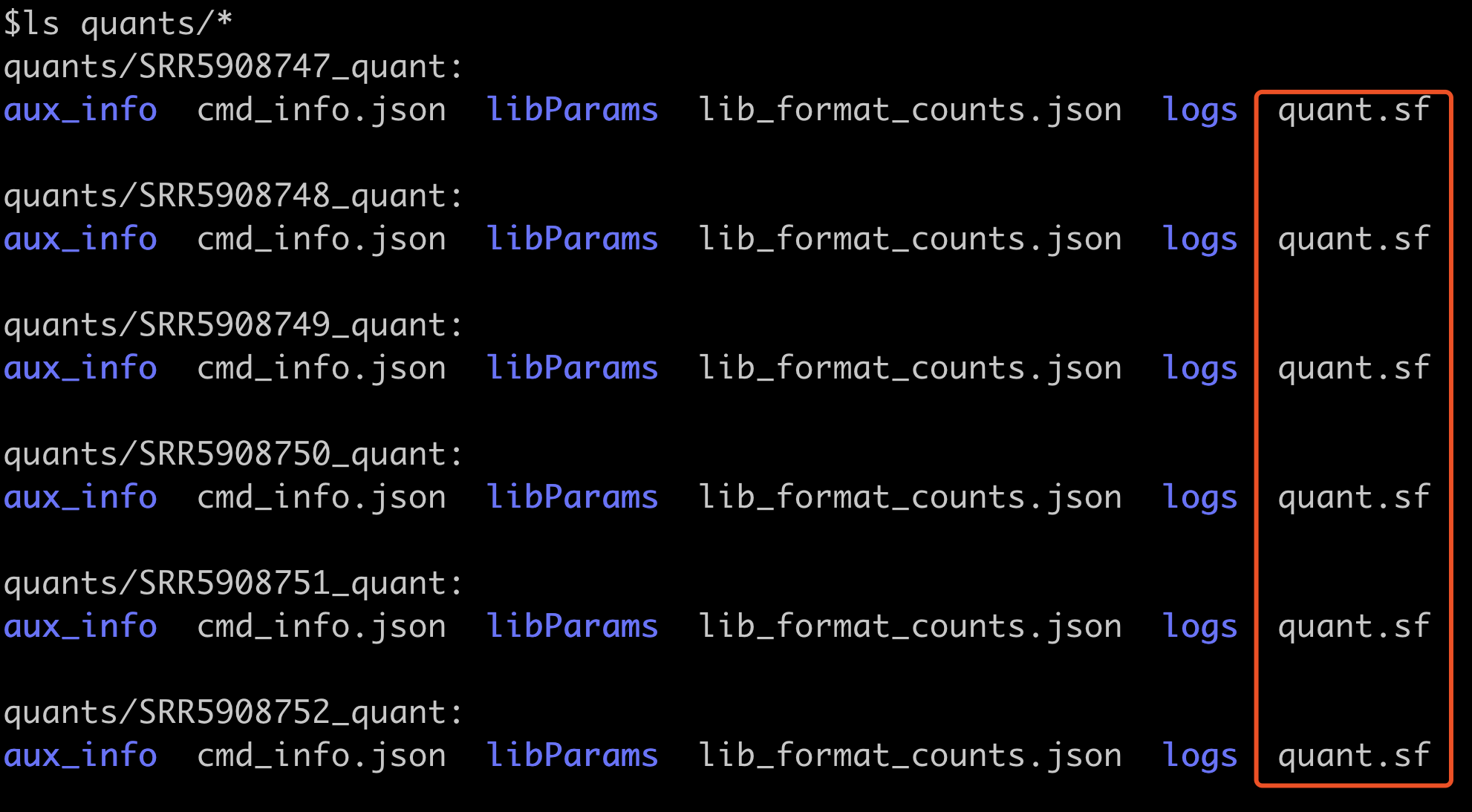
然后把所有的样本的quant.sf文件批量读取到R里面:
rm(list = ls())
options(stringsAsFactors = F)
library(rnaseqDTU)
library(tximport)
files=list.files('quants/',pattern = 'quant.sf',recursive = T,full.names = T)
txi <- tximport(files, type="salmon", txOut=TRUE,
countsFromAbundance="scaledTPM")
cts <- txi$counts
cts <- cts[rowSums(cts) > 0,]
library(GenomicFeatures)
# 文件 gencode.v32.annotation.gtf.gz 自己在gencode数据库网页下载即可
gtf <- "../database/gencode/gencode.v32.annotation.gtf.gz"
txdb.filename <- "gencode.v32.annotation.sqlite"
txdb <- makeTxDbFromGFF(gtf)
saveDb(txdb, txdb.filename)
txdb <- loadDb(txdb.filename)
txdf <- select(txdb, keys(txdb, "GENEID"), "TXNAME", "GENEID")
tab <- table(txdf$GENEID)
txdf$ntx <- tab[match(txdf$GENEID, names(tab))]
cts[1:3,1:3]
range(colSums(cts)/1e6)
head(txdf)
cts=cts[rownames(cts) %in% txdf$TXNAME,]
dim(cts)
txdf <- txdf[match(rownames(cts),txdf$TXNAME),]
all(rownames(cts) == txdf$TXNAME)
counts <- data.frame(gene_id=txdf$GENEID,
feature_id=txdf$TXNAME,
cts)
save(counts,files,file = 'salmon-out.Rdata')
上面整理salmon结果的代码,看起来很复杂,其实修改的地方不多,值得注意的是:
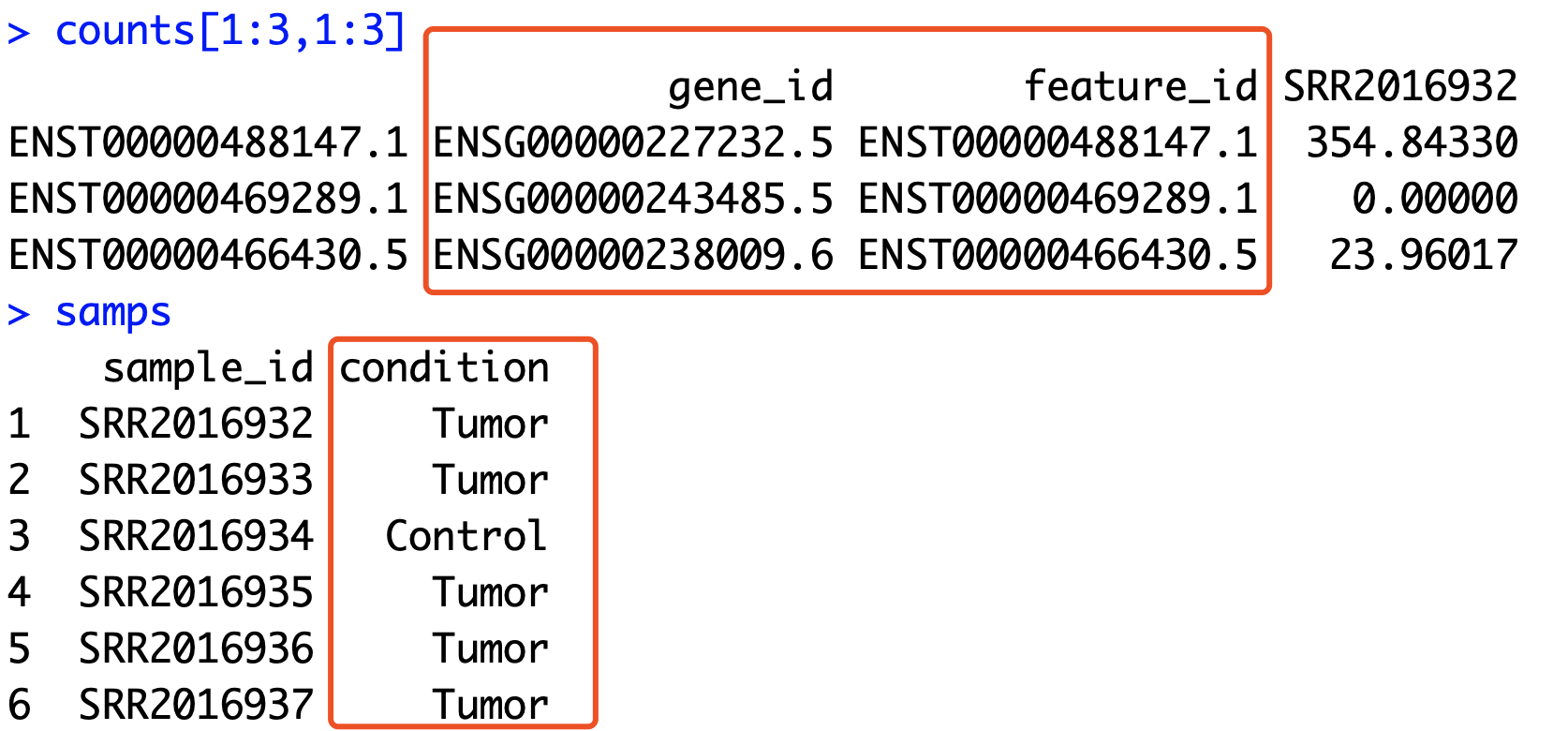
表达矩阵的第一列是基因的ID,第二列是转录本的ID,后面才是表达量哦。
有一个分组信息,我这里并没有给出我的代码,因为每个人的项目不一样,你需要自己制作,但凡有点R语言基础的,都是没有问题啦。就是 samps 那个变量的内容,有了它,下面的DRIMSeq流程分析差异转录本表达量才有意义。
接着运行DRIMSeq流程即可:
library(DRIMSeq)
d <- dmDSdata(counts=counts, samples=samps)
d
counts(d[1,])[,1:4]
n <- 12
n.small <- 6
d <- dmFilter(d,
min_samps_feature_expr=n.small, min_feature_expr=10,
min_samps_feature_prop=n.small, min_feature_prop=0.1,
min_samps_gene_expr=n, min_gene_expr=10)
d
table(table(counts(d)$gene_id))
design_full <- model.matrix(~condition, data=DRIMSeq::samples(d))
colnames(design_full)
table(samps$condition)
set.seed(1)
system.time({
d <- dmPrecision(d, design=design_full)
d <- dmFit(d, design=design_full)
d <- dmTest(d, coef="conditionControl")
})
res <- DRIMSeq::results(d)
head(res)
res.txp <- DRIMSeq::results(d, level="feature")
head(res.txp)
no.na <- function(x) ifelse(is.na(x), 1, x)
res$pvalue <- no.na(res$pvalue)
res.txp$pvalue <- no.na(res.txp$pvalue)
save(d,res,res.txp,file = 'DRIMSeq-out.Rdata')
是不是非常简单,就拿到了全部的转录本水平的差异表达呢,还可以可视化如下:
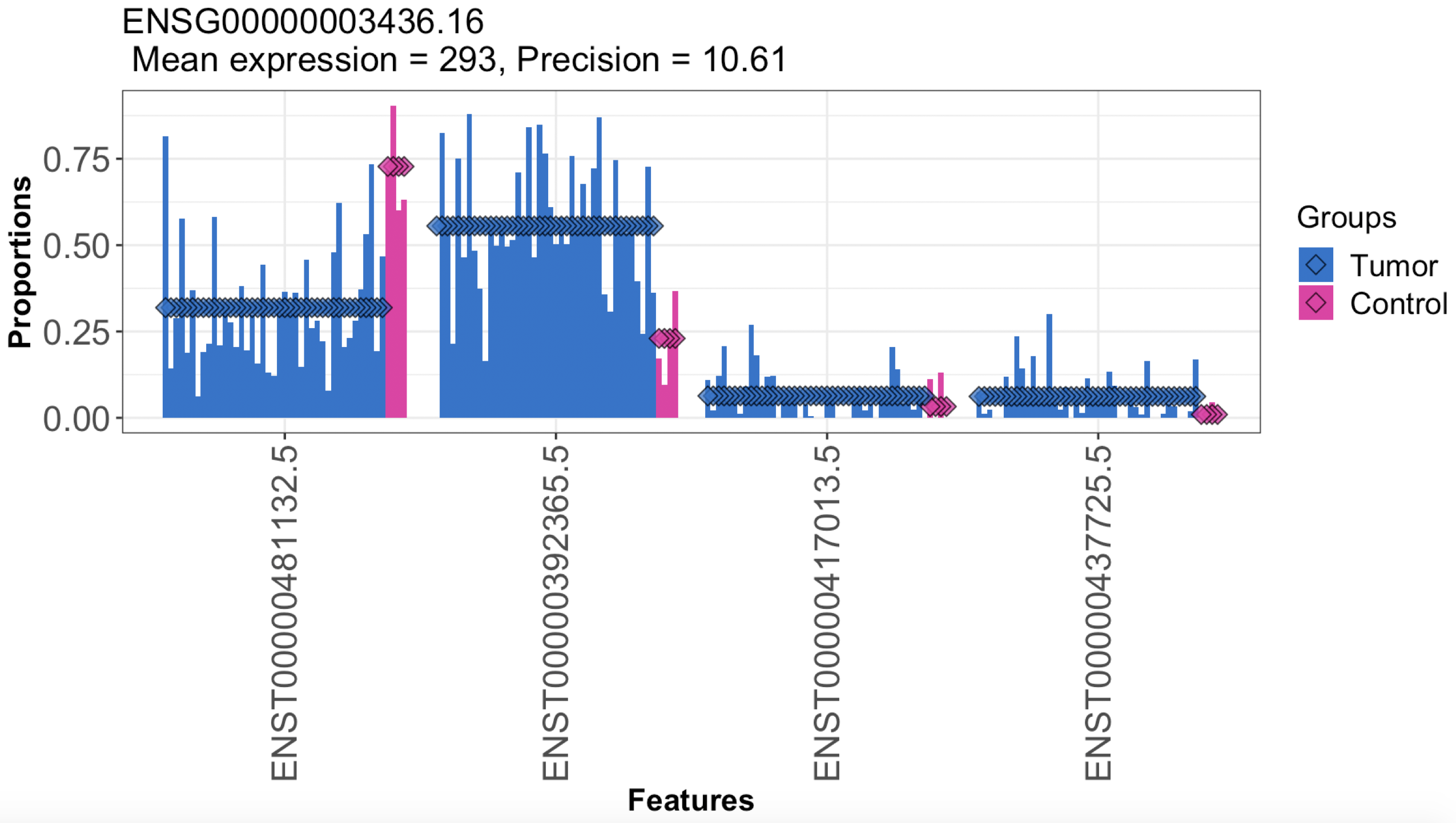
可以看到,我举例的这个项目里面,tumor和control组的样本量是不平衡的,而且基因ID也不容易理解,大家可以自行转换为基因的symbol,这样出图更直观。
学习这样的流程是需要一定背景知识的
首先是LINUX学习
我在《生信分析人员如何系统入门Linux(2019更新版)》把Linux的学习过程分成6个阶段 ,提到过每个阶段都需要至少一天以上的学习:
- 第1阶段:把linux系统玩得跟Windows或者MacOS那样的桌面操作系统一样顺畅,主要目的就是去可视化,熟悉黑白命令行界面,可以仅仅以键盘交互模式完成常规文件夹及文件管理工作。
- 第2阶段:做到文本文件的表格化处理,类似于以键盘交互模式完成Excel表格的排序、计数、筛选、去冗余,查找,切割,替换,合并,补齐,熟练掌握awk,sed,grep这文本处理的三驾马车。
- 第3阶段:元字符,通配符及shell中的各种扩展,从此linux操作不在神秘!
- 第4阶段:高级目录管理:软硬链接,绝对路径和相对路径,环境变量
- 第5阶段:任务提交及批处理,脚本编写解放你的双手
- 第6阶段:软件安装及conda管理,让linux系统实用性放飞自我
然后是R学习
我在生信分析人员如何系统入门R(2019更新版) 里面给初学者的知识点路线图如下:
- 了解常量和变量概念
- 加减乘除等运算(计算器)
- 多种数据类型(数值,字符,逻辑,因子)
- 多种数据结构(向量,矩阵,数组,数据框,列表)
- 文件读取和写出
- 简单统计可视化
- 无限量函数学习
必备书籍及视频
书籍贪多不烂,下面2本必买,读5遍以上
视频必须强推生信技能树近30万学习量的基础合辑:
生信技能树可变剪切相关教程节选
因为做目录确实很浪费时间,差不多就下面这些,大家先学习吧:
- 100篇泛癌研究文献解读之可变剪切事件大起底
- rMATS这款差异可变剪切分析软件的使用体验
- 用LeafCutter探索转录组数据的可变剪切
- 用Expedition来分析单细胞转录组数据的可变剪切
- 使用SGSeq探索可变剪切
- 超2万样本的RNA-seq数据重新统一处理(TCGA+GTEx+ TARGET)
- 玩转RNA-seq数据也可以不需要linux ?
- 高表达的PVT1(lncRNA)能够独立且有效地预测葡萄膜黑色素瘤生存情况
- RNA-seq技术已经常规化,你还好意思不掌握吗?
- 你值得拥有的单细胞RNA测序分析工具TOP 3
- 100篇泛癌研究文献解读之snoRNAs
- 100篇泛癌研究文献解读之lincRNA的生存分析情况
- 100篇泛癌研究文献解读之驱动lncRNA
- KM生存曲线经logRNA检验后也可以计算HR值
- 为什么我一行代码就可以完成3个R包的RNA-seq差异分析呢
- 玩转RNA-seq数据也可以不需要linux ?
- 你希望这个探针注释到蛋白编码基因还是miRNA的基因呢
- 如果miRNA的3p和5p功能不一样
- 哪怕是到了2018年,RNA-seq仍然可以不做重复
- 原创10000+生信教程大神给你的RNA实战视频演练
- TCGA的28篇教程-数据挖掘三板斧之ceRNA
- scRNA-seq的表达矩阵待解决的发育生物学问题
- 数据整理这一块工作商业公司可能做得更好-人类lncRNA大全
- GEO数据挖掘-第六期-RNA-seq数据也照挖不误
- 计算MiRNA–mRNA表达相关性
- 使用多个网页工具预测MiRNA–mRNA相互作用
- mRNA-seq数据中的duplicate情况探究
- Bioconductor包chimeraviz嵌合RNA可视化
- TCGA中GBM的RNA-seq和甲基化数据整合分析实践
- 四个公共scRNA-seq来测试算法
- lncRNA数据分析传送门
- RNA测序究竟有多可靠呢
- TCGA数据库里面的乳腺癌样本RNA-seq数据是配对的有哪些?
- 生信小白的RNA-seq实战历程
- RNA-seq数据分析指南
后记
听说隔壁openbiox团队在组织翻译这个bioconductor流程系列,而且还是由我们生信技能树元老-思考问题的熊领头,希望他们的翻译成果早日出版!