数据库构建也是生物信息学领域一个大方向,尤其是现在大热的单细胞领域,应该是不少团队在为单细胞数据库资源网页在踌躇满志了,不过单细胞数据之大,绝大部分实验室课题组是hold不住这个方向的数据这里的,最近看的一个预印本文章是:A curated database reveals trends in single-cell transcriptomics
- https://www.biorxiv.org/content/early/2019/10/17/742304.full.pdf
- The curated database described here is hosted at www.nxn.se/single-cell-studies.
- Forms for these functions are hosted at www.nxn.se/single-cell-studies/gui and www.nxn.se/single-cell-studies/submit.
不过我感兴趣的并不是他们做的单细胞资源整理,尽管他们收集了超过500个单细胞转录组研究的数据,我感兴趣的是他们文末的一个补充结论:
- Additionally, the database contains summarized information about analysis in the papers, allowing for analysis of trends in the field.
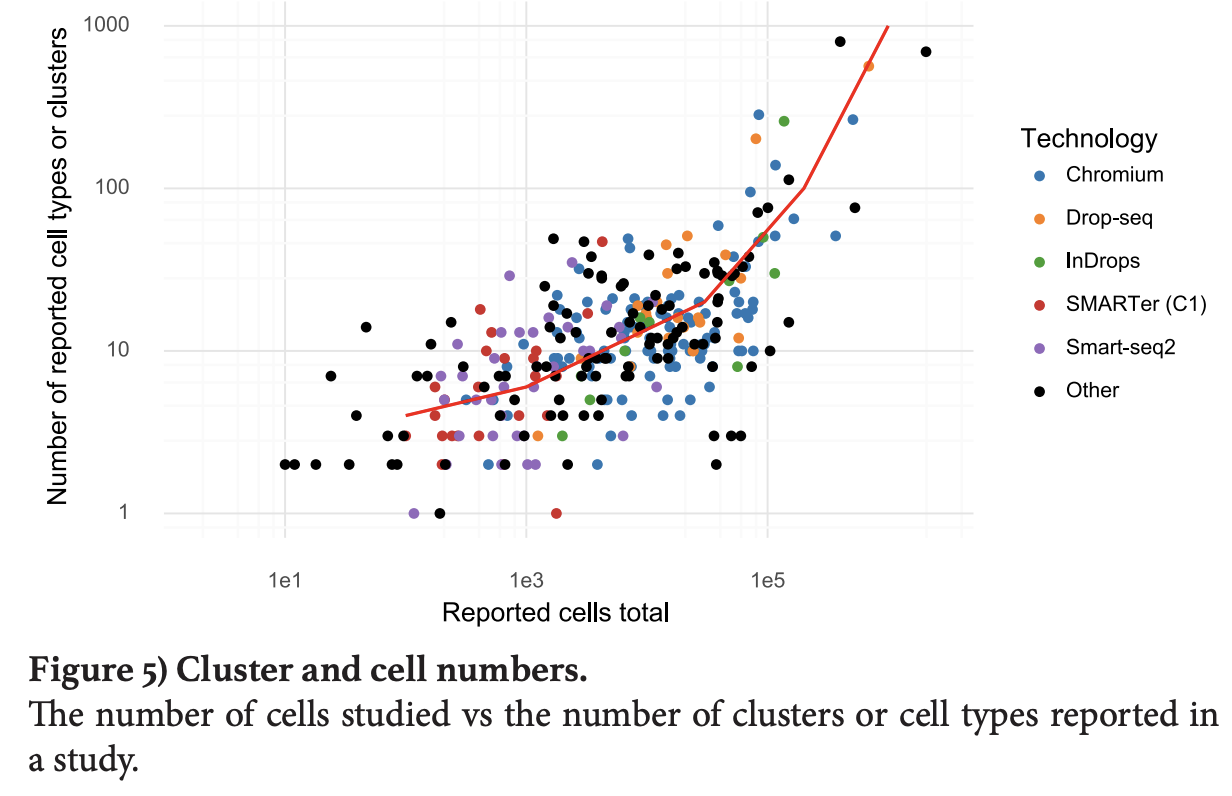
- As an example, we show that the number of cell types identified in scRNA-seq studies is proportional to the number of cells analysed.
就是说,不同的单细胞技术能检测到的细胞数量不一样,不同的技术有量级的差异,不同的数据分析方法也是会有不同的细胞亚群数量,但通常是没有量级的差异。不过,在整理这500个使用不同单细胞转录组技术的文章的分析结果发现一个很有趣的现象:检测到的单细胞数量和能分的细胞亚群数量是正比例相关的,如下所示:
这些不同的单细胞转录组技术我们在单细胞天地已经多次介绍过了,而且数据分析方法也公布了,如果是10X仪器的单细胞转录组数据走cellranger流程,我们在单细胞天地多次分享过流程笔记,大家可以自行前往学习,如下:
- 单细胞实战(一)数据下载
- 单细胞实战(二) cell ranger使用前注意事项
- 单细胞实战(三) Cell Ranger使用初探
- 单细胞实战(四) Cell Ranger流程概览
- 单细胞实战(五) 理解cellranger count的结果
如果是smart-seq2技术实际上是可以的啊!。不要仅仅是走单细胞下游分析标准流程啊,就是那些R包的认知,包括 scater,monocle,Seurat,scran,M3Drop 需要熟练掌握它们的对象,:一些单细胞转录组R包的对象 ,分析流程也大同小异:
- step1: 创建对象
- step2: 质量控制
- step3: 表达量的标准化和归一化
- step4: 去除干扰因素(多个样本整合)
- step5: 判断重要的基因
- step6: 多种降维算法
- step7: 可视化降维结果
- step8: 多种聚类算法
- step9: 聚类后找每个细胞亚群的标志基因
- step10: 继续分类
你可以做的更好!