看到九月份学徒在群里提问,写爬虫批量循环抓取NBCI数据库的基因信息,但是经常掉线,还有可能被封,求助!
我简单指点了他去找基因数据库文件即可,随便邀请他总结投稿如下:
一大早师姐给了个小任务,让我帮忙给注释下一批基因,格式类似如下:
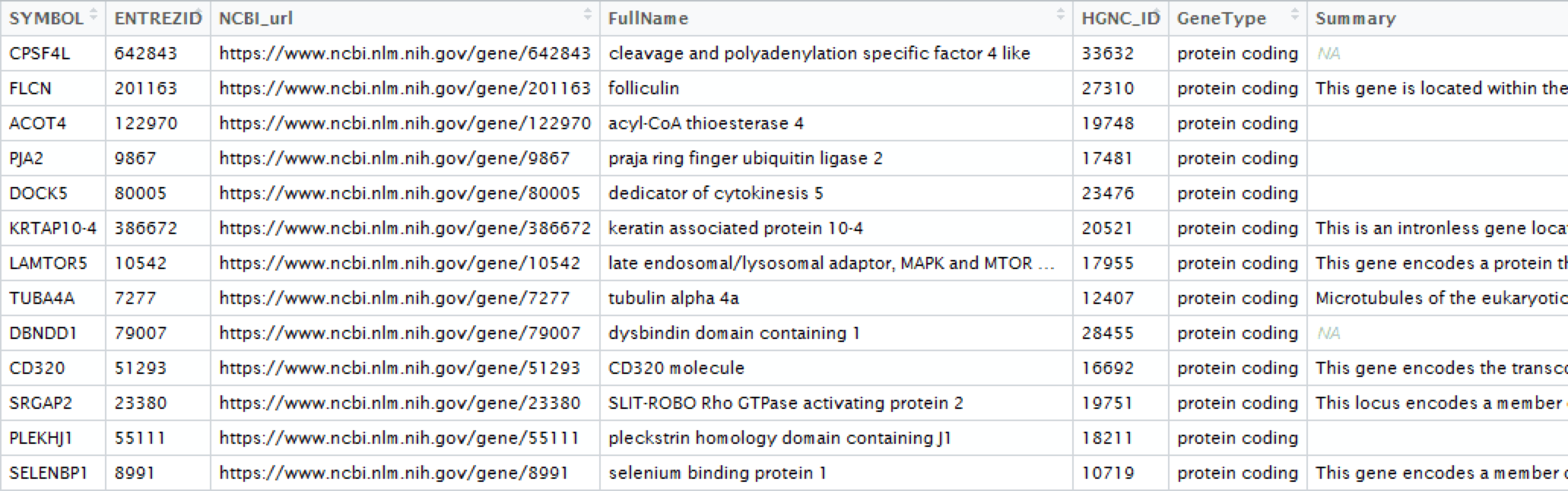
问了具体后原来是ncbi上的信息,相当于在ncbi上在gene库中查找,然后爬取目标信息。如下:
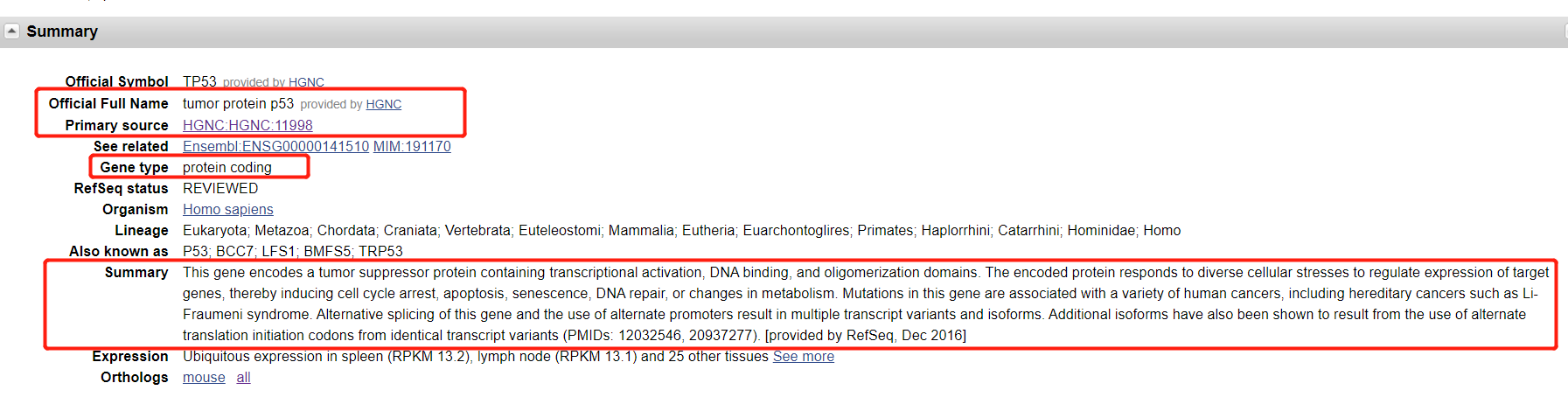
解决方案1:
我的第一反映就是用python爬虫去爬,想倒是挺好想的,但是太久没用python了,语法都忘得差不多了,于是就考虑使用R语言来做:
代码如下(如果可以翻墙这种方法也可行):
#install.packages("RCurl")
#install.packages("XML")
library(RCurl)
library(stringr)
library(XML)
library(clusterProfiler)
rm(list=ls())
options(stringsAsFactors = F)
# 读入基因列表:
genes <- read.table("HSC_MPP1_BMvsoldBM.csv",sep = ",",header = T)[,1]
# 将gene symbol转为entrze ID:
genes <- bitr(genes, fromType="SYMBOL", toType="ENTREZID", OrgDb="org.Hs.eg.db")
# 网址数据框:
genes$NCBI_url <- paste("https://www.ncbi.nlm.nih.gov/gene/",genes$ENTREZID,sep="")
head(genes)
# 根据xpath获取节点内容:
getNodesTxt <- function(html_txt1,xpath_p){
els1 = getNodeSet(html_txt1, xpath_p)
# 获得Node的内容,并且去除空字符:
els1_txt <- sapply(els1,xmlValue)[!(sapply(els1,xmlValue)=="")]
# 去除\n:
str_replace_all(els1_txt,"(\\n )+","")
}
# 处理节点格式,为character且长度为0的赋值为NA:
dealNodeTxt <- function(NodeTxt){
ifelse(is.character(NodeTxt)==T && length(NodeTxt)!=0 , NodeTxt , NA)
}
# 简单使用xpath来获取:
for(i in 1:nrow(genes)){
# 获得网址:
doc <- getURL(genes[i,"NCBI_url"])
cat("成功获得网页!\t")
# 获得网页内容
html_txt1 = htmlParse(doc, asText = TRUE)
# 获得Full Name:
genes[i,"FullName"] <- dealNodeTxt(getNodesTxt(html_txt1,'//*[@id="summaryDl"]/dd[2]/text()'))
cat("写入基因\t")
# 获得HGNC ID:
genes[i,"HGNC_ID"] <- str_replace_all(dealNodeTxt(getNodesTxt(html_txt1,'//*[@id="summaryDl"]/dd[3]/a')),"HGNC|:","")
cat("写入HGNC_ID\t")
# 获得Gene type:
genes[i,"GeneType"] <- dealNodeTxt(getNodesTxt(html_txt1,'//*[@id="summaryDl"]/dd[5]/text()'))
cat("写入GeneType\t")
# 获得summary:
genes[i,"Summary"] <- ifelse(length(getNodesTxt(html_txt1,'//*[@id="summaryDl"]/dd[10]/text()'))!=0,getNodesTxt(html_txt1,'//*[@id="summaryDl"]/dd[10]/text()'),NA)
cat("写入Summary\n")
print(paste("完成第",i,"个了!"))
}
可是,还没爬几条数据就出现了问题:因为网络问题,一是过快访问,存在被封ip的危险,二是访问ncbi如果不翻墙就很卡,甚至链接中断(使用了vpn后的确会有好转,但是不知道为什么vpn隔几分钟就中断,放弃了,太折腾了)
解决方案2
于是咨询了下Jimmy老师,Jimmy说有一种文件就可以直接有这些信息,于是乎:
发朋友圈求助各路大神!!!
于是还是有大神来帮助的:

特别感谢!
于是,思路转变。
进入 http://www.ensembl.org/index.html 官网
找到目的界面:
可以看到里面有很多选项可以自己设置
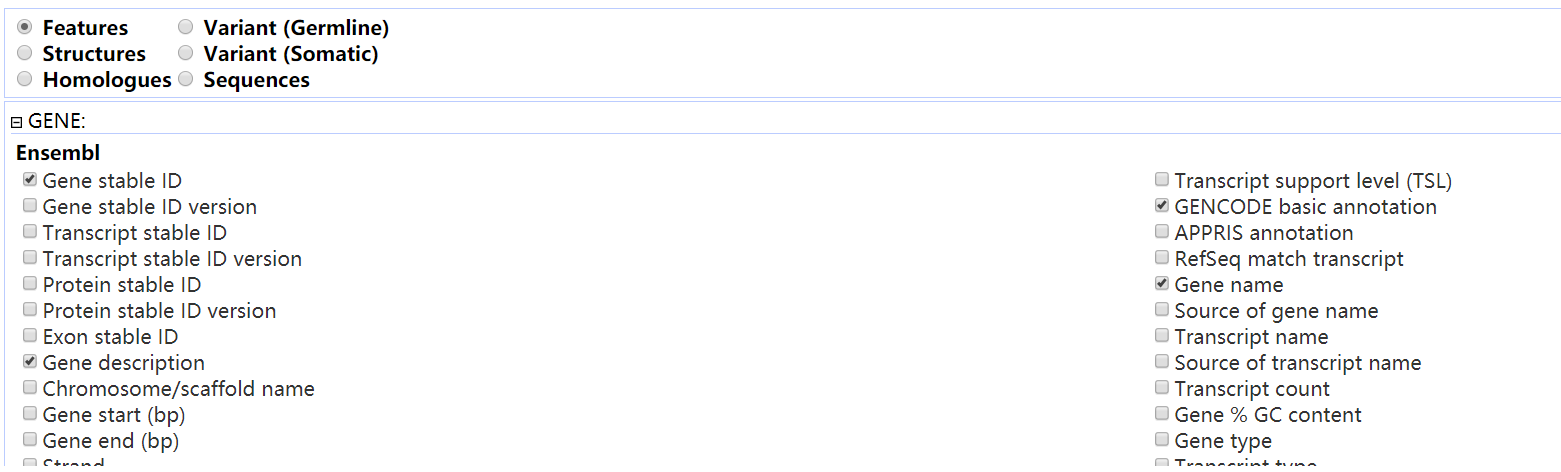
根据自己的需求自行设置吧:
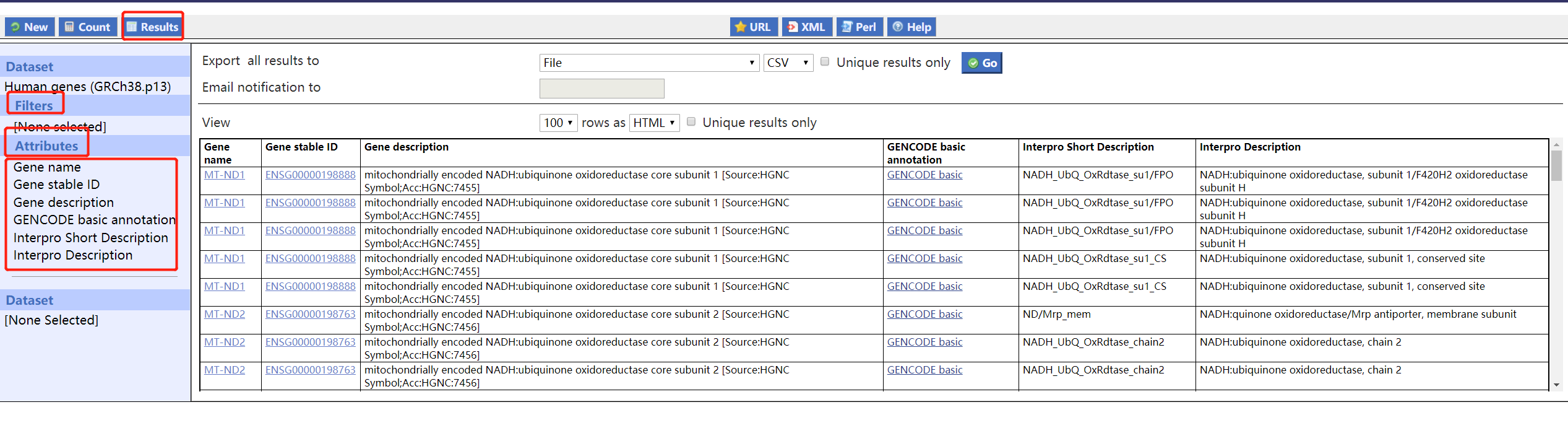
点击go下载文件,打开后如下:
基本上有点像了。于是导入R中进行后续处理,后面就很简单了,大家可以根据自己的需求自己去设置了。
还是贴上我写的垃圾代码,大家看看就好,别评论,丢不起这个人。
rm(list=ls())
options(stringsAsFactors = F)
# 读入基因列表:
genes <- read.table("HSC_MPP1_BMvsoldBM.csv",sep = ",",header = T)[,1]
tmpdb <- read.delim("mart_export.txt",sep = ",",header = T)
tmp=tmpdb[,c(1:3,5,6)]
#去除重复
tmp=tmp[!duplicated(tmpdb[,1]),]
target=tmp[match(genes,tmp[,1]),]
all <- read.table("HSC_MPP1_BMvsoldBM.csv",sep = ",",header = T)
all=all[,c(1,2,6,3)]
colnames(all)
colnames(tmp)
target=merge(tmp,all,by.y = "X",by.x = "Gene.name")
head(target)
target=target[,c(1,6:8,3:5)]
target=target[order(target$p_val_adj),]
write.table(target,file = "gene_annotation.xls",sep = "\t",row.names = F,col.names = T)
最后生成的文件如下:
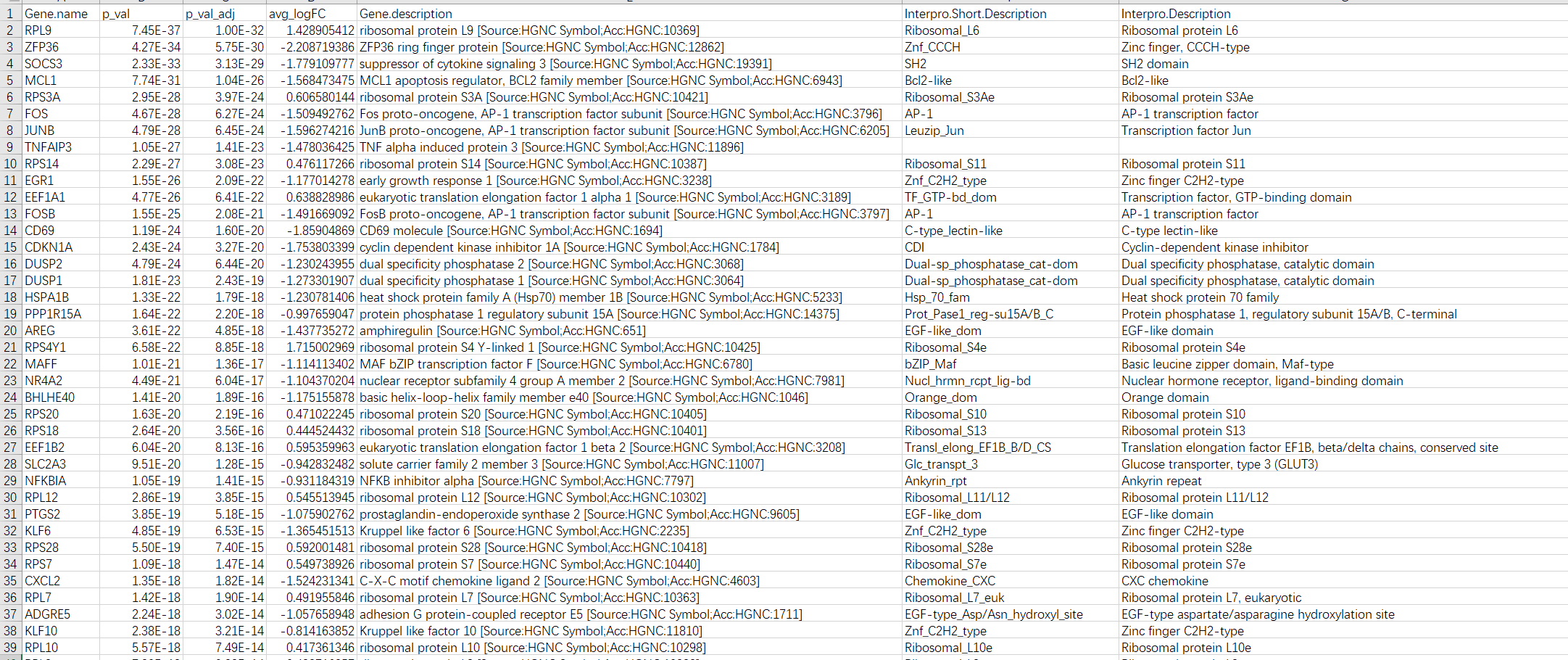
可以,很棒,就这样了,滚去复习六级了。