gtf文件大家都了解,基因或者外显子的坐标相对独立,但是转录本很不一样,同一个基因的不同转录本共用外显子,这样的话它们的坐标其实很多都是overlap的,这样,我们的二代测序的100bp或者150bp的reads就无法判定它到底属于哪一个转录本!(这个时候全长转录组测序(iso-seq)可能是更好的选择)
比如文章: 2015 Nov 7. doi: 10.1080/01621459.2015.1040880 就尝试图解如下:
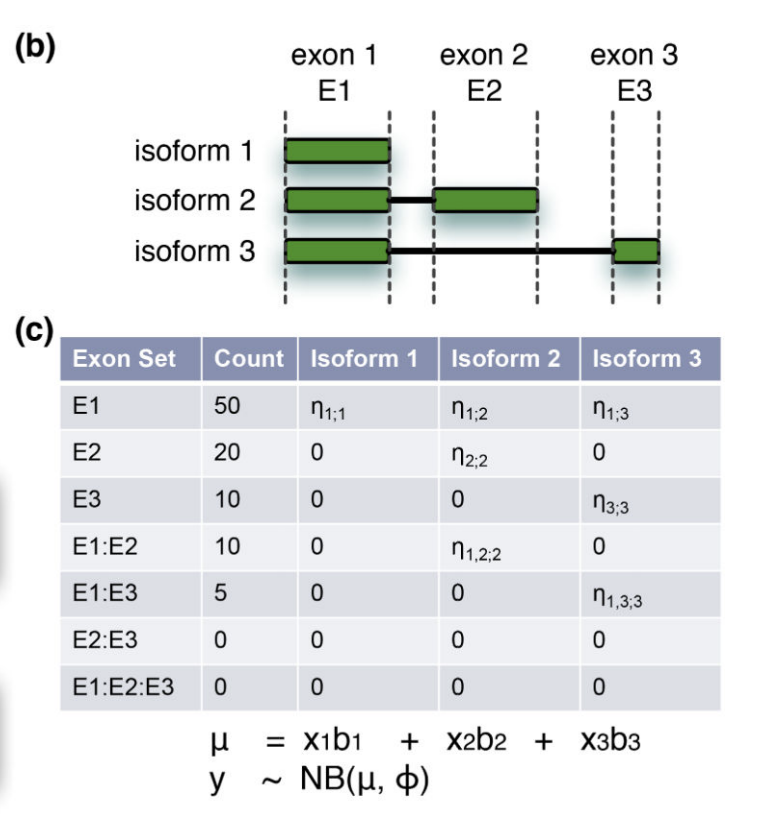
我们可以很轻松的对这个基因的3个exon进行表达量计数,但是呢,这3个转录本就需要通过公式来推断了,而且这还是一种理想的情况下,我们的这个基因仅仅是有这3个转录本,所以得到这样的结果,就不奇怪了。
TCGA数据库也不提供基于转录本的表达矩阵
比如我一直强推的UCSC的XENA数据库里面:
https://xenabrowser.net/datapages/
比如对BRCA来说,基于exon的表达矩阵是:
https://tcga.xenahubs.net/download/TCGA.BRCA.sampleMap/HiSeqV2_exon.gz
Level_3 data (file names: *.exon_quantification.txt) are downloaded from TCGA DCC, log2(x+1) transformed, and processed at UCSC into Xena repository.
input data formatROWs (identifiers) x COLUMNs (samples) (i.e. genomicMatrix)
239,323 identifiers X 1218 samples
基于基因的表达矩阵如下:
https://tcga.xenahubs.net/download/TCGA.BRCA.sampleMap/HiSeqV2.gz
Level_3 data (file names: *.rsem.genes.normalized_results) are downloaded from TCGA DCC, log2(x+1) transformed, and processed at UCSC into Xena repository
input data formatROWs (identifiers) x COLUMNs (samples) (i.e. genomicMatrix)
20,531 identifiers X 1218 samples
不过还好有专门的isoform数据库
我们下次再讲。