表达矩阵的标准分析通常是不够的,定位到成百上千个有统计学显著变化的差异表达基因后,同样是可以有成百上千个生物学功能注释(最出名的是GO功能和KEGG通路),普通的超几何分布检验已经不能满足大家多元化的分析了,所以就有了大家耳熟能详的GSEA分析,以及绝大部分人比较陌生的GSVA分析。
GSVA分析的文章发表于2013年,GSVA: gene set variation analysis for microarray and RNA-Seq data 同样是broad 研究生出品,其在2005年PNAS发表的gsea已经高达1.4万的引用了,不过这个GSVA才不到300。去年我就介绍过一波它的分析流程,在:使用GSVA方法计算某基因集在各个样本的表现 非常简单的代码,所以各个培训机构,公司人员都开始学习和二次创作进而分享。
成百上千个生物学功能注释(最出名的是GO功能和KEGG通路)通常是从数据库里面下载,就是基因集,也可以叫pathway,如下:
pathway的具体含义
pathway在我这里是基因集的别名,其中msigdb有着丰富的基因集,MSigDB(Molecular Signatures Database)数据库中定义了已知的基因集合:http://software.broadinstitute.org/gsea/msigdb 包括H和C1-C7八个系列(Collection),每个系列分别是:
- H: hallmark gene sets (癌症)特征基因集合,共50组,最常用;
- C1: positional gene sets 位置基因集合,根据染色体位置,共326个,用的很少;
- C2: curated gene sets:(专家)校验基因集合,基于通路、文献等:
- C3: motif gene sets:模式基因集合,主要包括microRNA和转录因子靶基因两部分
- C4: computational gene sets:计算基因集合,通过挖掘癌症相关芯片数据定义的基因集合;
- C5: GO gene sets:Gene Ontology 基因本体论,包括BP(生物学过程biological process,细胞原件cellular component和分子功能molecular function三部分)
- C6: oncogenic signatures:癌症特征基因集合,大部分来源于NCBI GEO 发表芯片数据
- C7: immunologic signatures: 免疫相关基因集合。
但是是可以自定义基因集
之所以大家不知道可以自定义基因集,其实是因为,大家做数据分析的时候,习惯了软件包作者打包或者说封装好的函数,如下:
library(clusterProfiler)
data(gcSample)
enrichKK <- enrichKEGG(gcSample[[1]])
enrichKK
class(enrichKK)
简单的几句话就对你感兴趣的基因集,进行了KEGG数据库的普通的超几何分布检验,是不是很神奇,你压根就没有去下载KEGG数据库,也不知道里面有多少条通路,每个通路具体多少个基因,不同通路之间基因的重合情况。
实际上我给大家写的GEO数据库挖掘的标准代码教程里面是有GSEA分析的:
library(ggplot2)
library(clusterProfiler)
library(org.Hs.eg.db)
### 对 MigDB中的全部基因集 做GSEA分析。
# http://www.bio-info-trainee.com/2105.html
# http://www.bio-info-trainee.com/2102.html
{
load(file = 'anno_DEG.Rdata')
geneList=DEG$logFC
names(geneList)=DEG$symbol
geneList=sort(geneList,decreasing = T)
#选择gmt文件(MigDB中的全部基因集)
d='../MsigDB/symbols'
gmts <- list.files(d,pattern = 'all')
gmts
#GSEA分析
library(GSEABase) # BiocManager::install('GSEABase')
## 下面使用lapply循环读取每个gmt文件,并且进行GSEA分析
## 如果存在之前分析后保存的结果文件,就不需要重复进行GSEA分析。
f='gsea_results.Rdata'
if(!file.exists(f)){
gsea_results <- lapply(gmts, function(gmtfile){
# gmtfile=gmts[2]
geneset <- read.gmt(file.path(d,gmtfile))
print(paste0('Now process the ',gmtfile))
egmt <- GSEA(geneList, TERM2GENE=geneset, verbose=FALSE)
head(egmt)
# gseaplot(egmt, geneSetID = rownames(egmt[1,]))
return(egmt)
})
# 上面的代码耗时,所以保存结果到本地文件
save(gsea_results,file = f)
}
load(file = f)
#提取gsea结果,熟悉这个对象
gsea_results_list<- lapply(gsea_results, function(x){
cat(paste(dim(x@result)),'\n')
x@result
})
gsea_results_df <- do.call(rbind, gsea_results_list)
gseaplot(gsea_results[[2]],'KEGG_CELL_CYCLE')
gseaplot(gsea_results[[2]],'FARMER_BREAST_CANCER_CLUSTER_6')
gseaplot(gsea_results[[5]],'GO_CONDENSED_CHROMOSOME_OUTER_KINETOCHORE')
}
看起来是稍微有点复杂了,其实如果你但凡是听过我的R语言课程,就很容易理解。核心就是2句话,读gmt格式的文件,通常是在,MSigDB(Molecular Signatures Database)数据库里面下载的文件,如下
然后做GSEA或者GSVA分析罢了,如果是GSEA,就:
library(GSEABase) # BiocManager::install('GSEABase')
geneset <- read.gmt(file.path(d,gmtfile))
egmt <- GSEA(geneList, TERM2GENE=geneset, verbose=FALSE)
head(egmt)
如果是GSVA,就:
library(GSVA) # BiocManager::install('GSVA')
geneset <- getGmt(file.path(d,gmtfile))
es.max <- gsva(X, geneset,
mx.diff=FALSE, verbose=FALSE,
parallel.sz=1)
可以看到,它们读取gmt格式文件的函数不一样,不过问题不大,只需要是标准的gmt格式文件即可,那么什么是标准的gmt格式文件呢,简单谷歌即可:
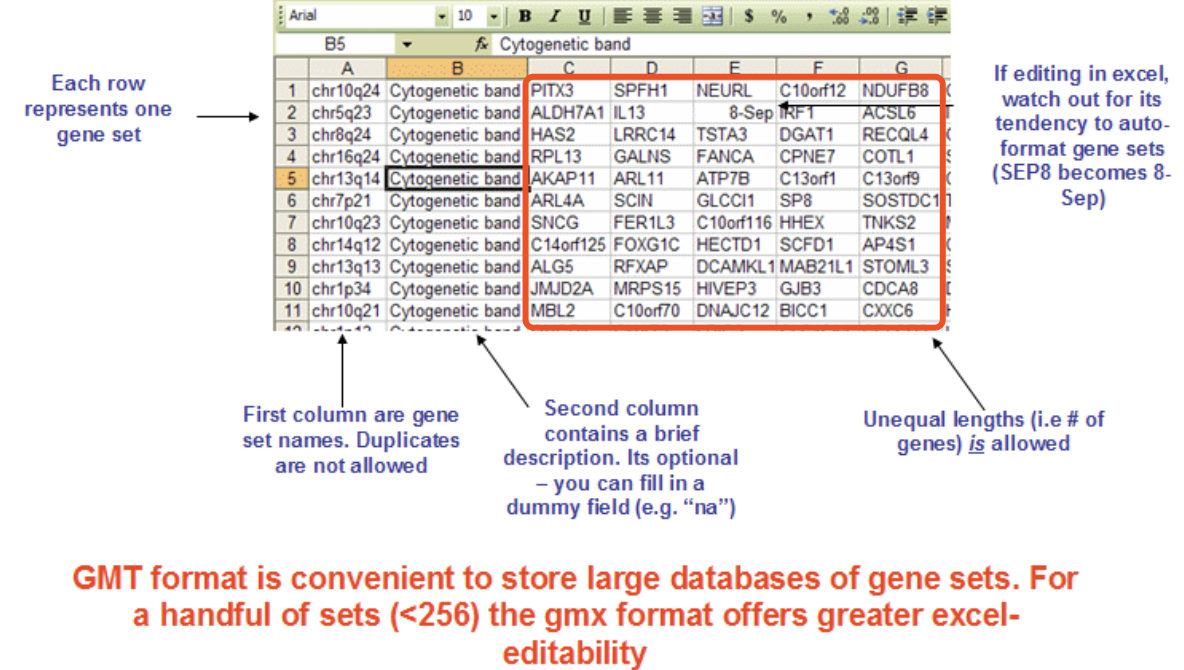
其实就是,每个基因集是一行,然后第一列是基因集的ID,第二列是名字,后面的列就是基因集所含有的基因。
而不同的基因集,在不同的行,可以有不同数量的基因啦。
所以你只需要自己制作这样的gmt文件,就可以啦,使用上面我们提到的函数进行读取。
如果你自己手动制作gmt文件困难肿么办
几年前我写过一个函数,专门把基因集,写出成为gmt文件,你可以试试看,能不能达到我几年前的水平哈!