关于RNA-seq,我们在生信技能树应该是至少推出了400篇教程,而且是我们全国巡讲的标准品知识点,其中还有一个阅读量过两万的综述翻译及其细节知识点的补充:
相信大家听完了我B站的RNA-seq分析流程后,对这个数据的应用方向都不陌生。最近连续收到好几个求助,都是关于转录组测序的counts矩阵去除批次效应,值得写推文解答一下咯!
首先安装R包pasilla
options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/")
options("repos" = c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
options()$repos
options()$BioC_mirror
BiocManager::install("pasilla",ask = F,update = F)
BiocManager::install("DESeq",ask = F,update = F)
简单了解
其实我们只需要使用表达矩阵和表型信息即可
library(pasilla)
data("pasillaGenes")
> pasillaGenes
CountDataSet (storageMode: environment)
assayData: 14470 features, 7 samples
element names: counts
protocolData: none
phenoData
sampleNames: treated1fb treated2fb ... untreated4fb (7 total)
varLabels: sizeFactor condition type
varMetadata: labelDescription
featureData: none
experimentData: use 'experimentData(object)'
pubMedIds: 20921232
Processed data from NCBI Gene Expression Omnibus under accession numbers GSM461176 to GSM461181.
cts=counts(pasillaGenes)
coldata=Biobase::pData(pasillaGenes)
coldata <- coldata[,c("condition","type")]
rownames(coldata) <- sub("fb", "", rownames(coldata))
coldata$type <- sub("-", "_", coldata$type)
coldata$type <- as.factor(coldata$type)
colnames(cts)=rownames(coldata)
coldata
cts[1:4,1:4]
表达矩阵和表型信息如下:
> coldata
condition type
treated1 treated single_read
treated2 treated paired_end
treated3 treated paired_end
untreated1 untreated single_read
untreated2 untreated single_read
untreated3 untreated paired_end
untreated4 untreated paired_end
> cts[1:4,1:4]
treated1 treated2 treated3 untreated1
FBgn0000003 0 0 1 0
FBgn0000008 78 46 43 47
FBgn0000014 2 0 0 0
FBgn0000015 1 0 1 0
>
很明显,我们关心的是treated和untreated两个组的差异,而单端测序和双端测序是我们想要抹去的批次效应,因为这篇文章是2010的数据,十年过去了,那个年代NGS是很稀罕的事情,测序仪还在不停的进化,单端测序和双端测序混在一起是容易理解的事情。
这个时候差异分析需要考虑批次效应
很多人以为去除批次效应是要改变你的表达矩阵,新的表达矩阵去走差异分析流程,其实大部分的差异分析流程包里面,人家内置好了考虑你的批次效应这样的混杂因素的函数用法设计,比如:
library(DESeq2)
dds <- DESeqDataSetFromMatrix(countData = cts,
colData = coldata,
design = ~ type+condition)
dds <- DESeq(dds)
resultsNames(dds)
res <- results(dds, name="condition_untreated_vs_treated")
summary(res)
做出来的差异分析结果是:
out of 11836 with nonzero total read count
adjusted p-value < 0.1
LFC > 0 (up) : 544, 4.6%
LFC < 0 (down) : 458, 3.9%
outliers [1] : 0, 0%
low counts [2] : 3629, 31%
(mean count < 5)
[1] see 'cooksCutoff' argument of ?results
[2] see 'independentFiltering' argument of ?results
可以拿它去绘制火山图,差异基因的热图等等下游分析。
因为我们把批次这个因素,写在了DESeq2里面,所以它在帮我们做差异分析的时候,其实就考虑得到的差异分析结果,就是去除了批次效应的。
如果你确实一定要亲眼看看批次效应到底是如何影响这个表达矩阵的,就需要看PCA啦。
使用limma的removeBatchEffect去除批次效应
知道注意的是limma的removeBatchEffect这个时候肯定会改变你的counts值矩阵,改变后就没办法走DESeq2差异分析流程啦,仅仅是为了拿到去除批次效应前后对比的表达矩阵而已。
library(ggplot2)
vsd <- vst(dds, blind=FALSE)
pcaData <- plotPCA(vsd, intgroup=c("condition", "type"), returnData=TRUE)
percentVar <- round(100 * attr(pcaData, "percentVar"))
ggplot(pcaData, aes(PC1, PC2, color=condition, shape = type)) +
geom_point(size=3) +
xlim(-12, 12) +
ylim(-10, 10) +
xlab(paste0("PC1: ",percentVar[1],"% variance")) +
ylab(paste0("PC2: ",percentVar[2],"% variance")) +
geom_text(aes(label=name),vjust=2)
ggsave("myPCAWithBatchEffect.png")
assay(vsd) <- limma::removeBatchEffect(assay(vsd), vsd$type)
pcaData <- plotPCA(vsd, intgroup=c("condition", "type"), returnData=TRUE)
percentVar <- round(100 * attr(pcaData, "percentVar"))
ggplot(pcaData, aes(PC1, PC2, color=condition, shape = type)) +
geom_point(size=3) +
xlim(-12, 12) +
ylim(-10, 10) +
xlab(paste0("PC1: ",percentVar[1],"% variance")) +
ylab(paste0("PC2: ",percentVar[2],"% variance")) +
geom_text(aes(label=name),vjust=2)
ggsave("myPCABatchEffectRemoved.png")
效果如下:
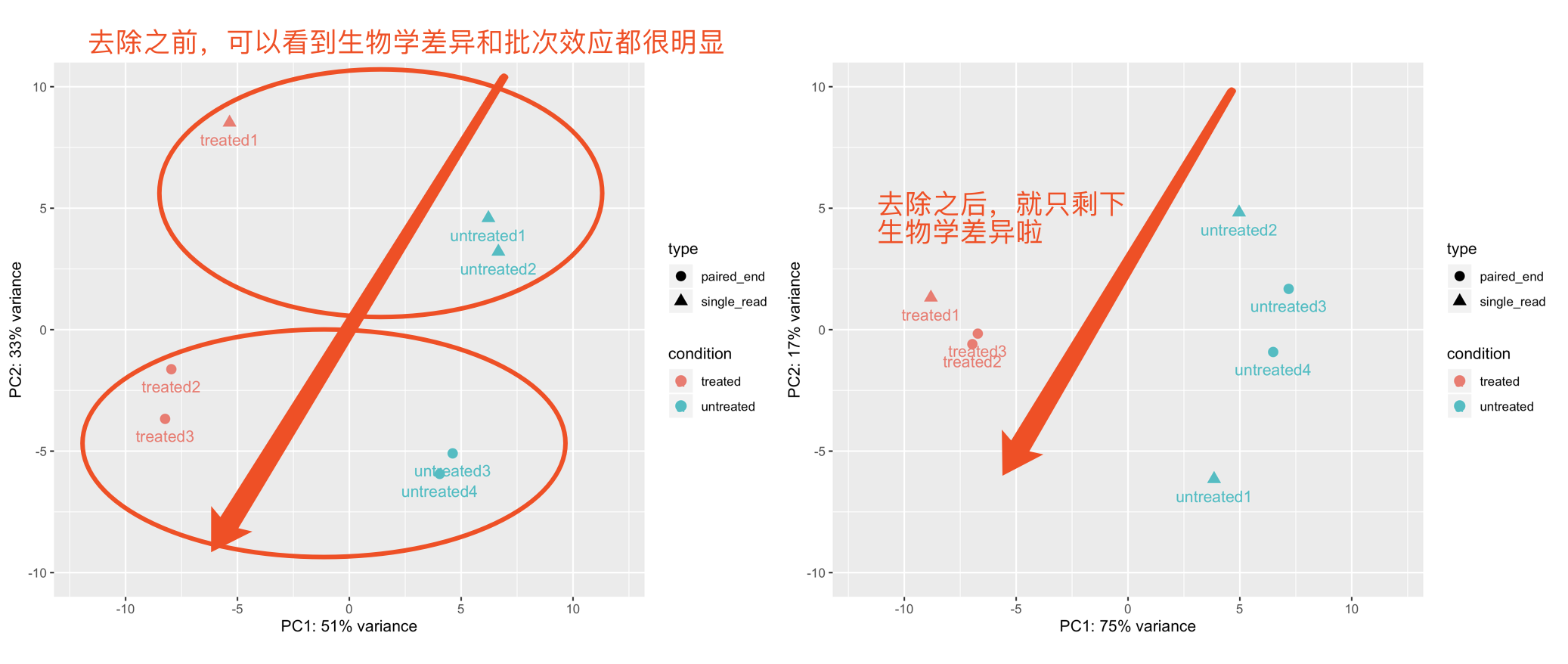
还可以绘制样本相关性热图啦:
library("pheatmap")
library("RColorBrewer")
sampleDists <- dist(t(assay(vsd)))
sampleDistMatrix <- as.matrix(sampleDists)
rownames(sampleDistMatrix) <- paste(vsd$condition, vsd$type, sep="-")
colnames(sampleDistMatrix) <- NULL
colors <- colorRampPalette( rev(brewer.pal(9, "Blues")) )(255)
pdf("heatmap.pdf", height = 4, width = 5)
pheatmap(sampleDistMatrix,
clustering_distance_rows=sampleDists,
clustering_distance_cols=sampleDists,
col=colors)
dev.off()
以上示例代码来源于:https://biohpc.cornell.edu/doc/RNA-Seq-2019-exercise3.html
但是我觉得这些并没有我自己的代码好:免费的数据分析付费的成品代码
总结一下(以下是付费内容)
如果你是要对具有批次效应的RNA-seq的count值矩阵进行差异分析,那么只需要你在DESeq2走差异分析流程的时候指明你的批次效应因素即可,无需改变矩阵。
如果你是要进行后续可视化呢,就需要改变矩阵啦,这个时候limma的removeBatchEffect是首选,需要注意的是它针对的是DESeq2里面vst之后的矩阵哦!(PS: 我在生信技能树分享过DESeq2里面vst函数,两年前,你可以自行搜索一下)
转录组测序的counts矩阵去除批次效应分析免费做
我们推文里面提到的各种各样的数据分析环节都是我非常有经验的,比如我在lncRNA的一些基础知识 ,和lncRNA芯片的一般分析流程 介绍过的那些图表,以及下面的目录的分析内容 对我来说是举手之劳,希望可以帮助到你!
- 转录组数据分析的4个维度认识(数据分析继续免费哦) RNA-seq数据的2个分组差异分析,热图,PCA图,火山图等等
- 根据感兴趣基因看肝癌免疫微环境的T细胞亚群差异 条形图或者箱线图
- 查看感兴趣基因的甲基化水平和RNA表达水平(数据分析免费做)相关性 散点图或者箱线图
- 我不相信kmplot这个网页工具的结果(生存分析免费做)
- 单基因GSEA分析策略(数据分析免费做活动继续)
- 干扰一个基因然后分析全局基因表达其实是无法定位该基因完整功能(春节免费数据分析活动继续)
- log与否会改变rpkm形式表达矩阵top的mad基因列表 WGCNA分析免费做
- 甲基化信号值的差异分析也许不应该是看logFC 甲基化信号矩阵差异分析免费做
- WGCNA得到模块之后如何筛选模块里面的hub基因 WGCNA分析免费做
- 既然可以看感兴趣基因的生存情况,当然就可以批量做完全部基因的生存分析
同样的,本次活动我可以帮你免费做一次转录组测序的counts矩阵去除批次效应分析,但是呢,我也没办法保证结果咋样,有时候数据集就是这样。而且,你需要挑选一下你的阈值哦!
还是老规矩,发送数据分析要求,以及简短的项目描述到我的邮箱 jmzeng1314@163.com
邮件正文最好是加上你是啥时候认识生信技能树的哦,或者其它一些寒暄的话,自我介绍也行。主要是考虑到可能想免费分析数据的朋友很多,所以会根据你的来信,我主观判定一个优先级哦。目前我有20多个愿意长期在我的指导下进行数据探索的学徒,等我的团队扩大到200人,我们应该是可以做到数据分析全部免费,敬请期待哈!