今天在群里看到一个提问,很有趣,好像是有一个网页工具可以把fastq格式转为phylip格式。我虽然没有使用过这个软件,但是我觉得这个提问,可能是忽略了计算过程,直接说结果。应该是问题本身就错误的。
我们四年前就系统性整理过生信基础知识名词本,其中就有数据格式专题,目录如下:
- 1.FastQ和FastA格式
- 2.SAM格式
- 3.gff/gtf格式
- 4.Bigwig/Wiggle格式
- 5.bed格式
- 6.vcf格式
- 7.Blast&Blat
虽然没有phylip格式,但是如果你理解了格式,就知道,其实无非就是软件开发者定义好的规则。我以前分享过HPV的病毒进化树,可以把这个当做是学徒作业了。https://bigd.big.ac.cn/ncov
国家生物信息中心
2019新型冠状病毒信息库 (2019nCoVR)
大机构大组织做的很全面:

不仅仅是有热点病毒序列,甚至有全部的冠状病毒信息,下面是网页公布的一些信息 - 新增来源于GenBank,GISAID的11条基因组序列。(2020.03.06)
- 对146条人源新冠病毒全基因组序列进行质控分析,发现有1条序列有多个Ns和简并碱基,2条序列有多个Gaps,2条序列的变异数量较多,6条序列的变异有密集分布区。
- 基于142条高质量人源新冠病毒全基因组序列变异分析,共鉴定224个变异(115个使氨基酸发生变化),根据群体发生率和变异分布进行变异分级(I-III,详见变异注释表)。
- 基于原始测序数据,对新冠病毒Wuhan-Hu-1毒株进行基因组拼接,序列比较和共线性分析显示两者序列完全一致,进一步确认该毒株基因组序列NC_045512.2的准确性。
- 2019新型冠状病毒信息库在《遗传》期刊在线发表。
- 由中国医学科学院病原生物学研究所提交的5株2019新型冠状病毒全基因组序列已经通过国家生物信息中心/国家基因组科学数据中心与NCBI共享,序列号为MT019529~MT019533。
机构对这些序列进行了一些探索,绘制了两个典型的图,如下:病毒基因组单倍型网络
这个图没有看出来是怎么制作的,多序列比对可能不够,看起来应该是固定一个病毒序列为参考基因组,然后所有的其它地方检测到的病毒也进行测序后,比对到我们人为规定的参考基因组上面,看变异位点哦。

一般来说,其它地方检测到的病毒基因组变异位点都是在个位数这样的数量级,因为病毒进化是有速度的。系统发生树
这个就比较常规了,如果只有Windows电脑的,使用mega就足够了
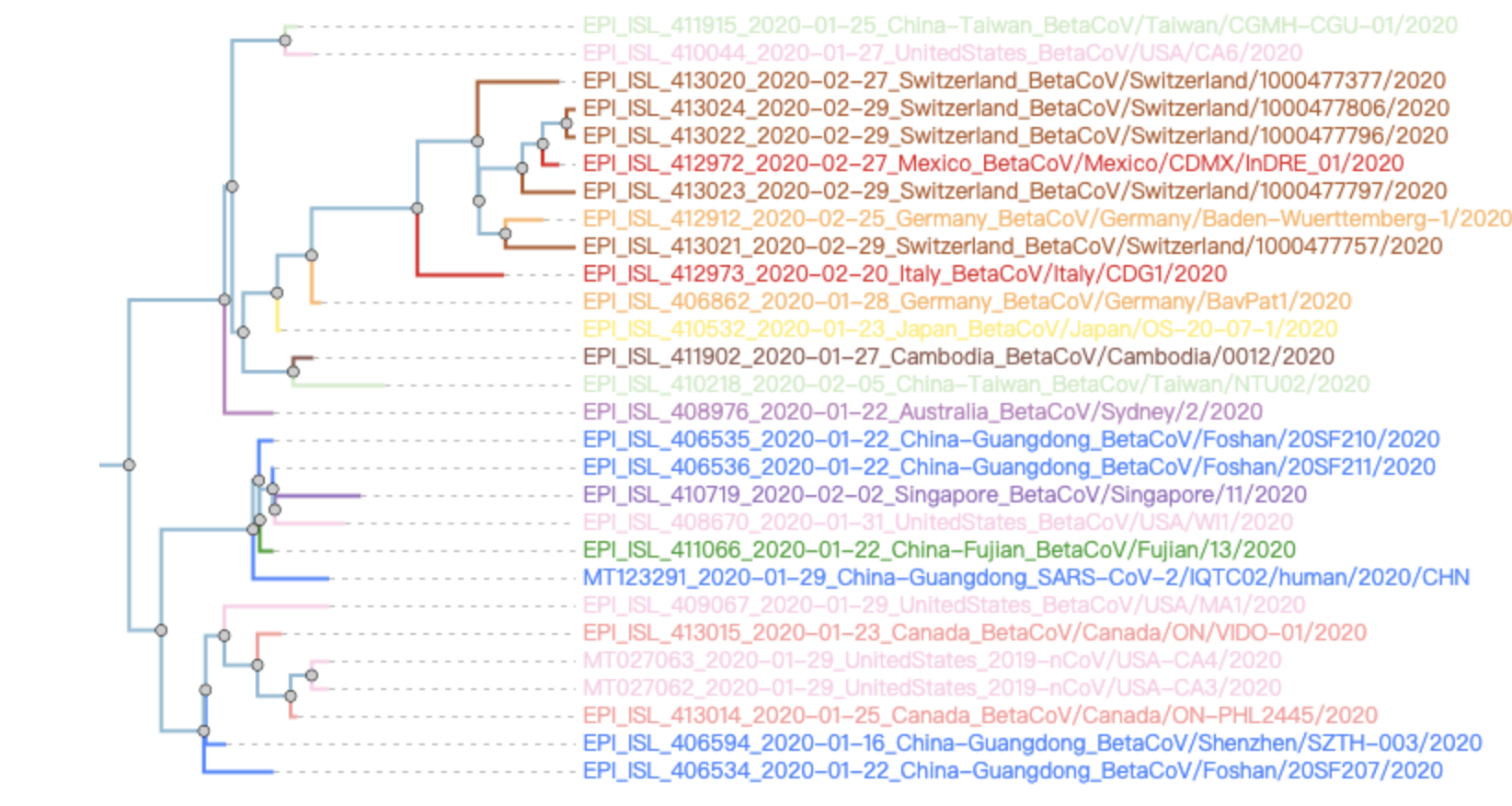
如果有Linux操作系统,可以参考我以前的博客: - http://www.bio-info-trainee.com/659.html Muscle进行多序列比对
- http://www.bio-info-trainee.com/626.html 用phyML对多重比对phy文件来构建进化树
因为确实不做这方面研究,这个探索性课题,就留给我的学徒吧!文末友情宣传
强烈建议你推荐给身边的博士后以及年轻生物学PI,多一点数据认知,让他们的科研上一个台阶:
- 全国巡讲全球听(买一得五),第二期 ,你的生物信息学入门课
- 生信技能树的2019年终总结 ,你的生物信息学成长宝藏
- 2020学习主旋律,B站74小时免费教学视频为你领路