虽然是生物学过程很多,但是免疫的重要性毋庸置疑,大家的肿瘤研究课题最后很喜欢定位到免疫这个话题。
计算肿瘤的免疫得分的软件算法不少,其中ESTIMATE是一个还算比较容易理解的,优秀的工具,但是我发现关于它的教程非常少,而且基本上都以我多年前在生信技能树分享教程为原型:使用ESTIMATE来对转录组表达数据根据stromal和immune细胞比例估算肿瘤纯度
ESTIMATE (Estimation of STromal and Immune cells in MAlignant Tumor tissues using Expression data)
虽然说直接落脚到ESTIMATE的数据挖掘文章不多:100篇泛癌研究文献解读之ESTIMATE算法计算肿瘤纯度 ,但是用到它的文章却不少哦,大家可以去谷歌文献搜索看看它被引用的次数,我前面提到的:学徒数据挖掘代码打包 里面就有一个实例。最近有粉丝提问,我们讲解的都是芯片表达矩阵,如果是RNA-seq的counts矩阵该如何使用ESTIMATE计算肿瘤的免疫得分,以及这样做的合理性?
如果是RNA-seq的counts矩阵要使用ESTIMATE
那非常简单,我可以把ESTIMATE用法,包装成为一个函数:
dat=log2(edgeR::cpm(exprSet)+1)
library(estimate)
estimate <- function(dat,pro){
input.f=paste0(pro,'_estimate_input.txt')
output.f=paste0(pro,'_estimate_gene.gct')
output.ds=paste0(pro,'_estimate_score.gct')
write.table(dat,file = input.f,sep = '\t',quote = F)
library(estimate)
filterCommonGenes(input.f=input.f,
output.f=output.f ,
id="GeneSymbol")
estimateScore(input.ds = output.f,
output.ds=output.ds,
platform="illumina") ## 注意这个platform参数
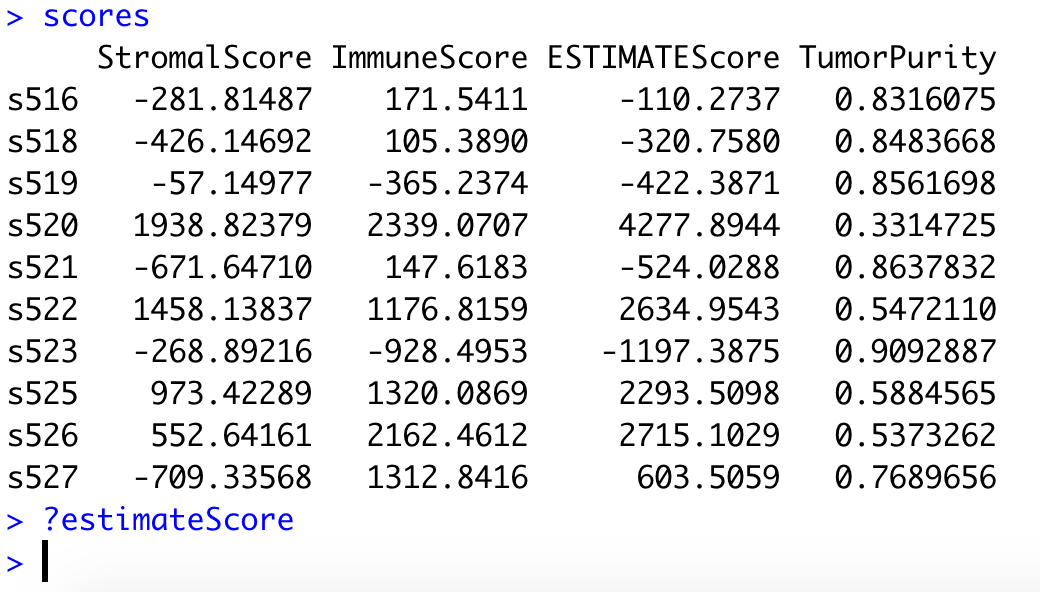
scores=read.table(output.ds,skip = 2,header = T)
rownames(scores)=scores[,1]
scores=t(scores[,3:ncol(scores)])
return(scores)
}
pro='bladder'
scores=estimate(dat,pro)
也就是说,你只需要给一个RNA-seq的counts矩阵,如上所示,我喜欢命名为exprSet,这样后续只需要使用我打包好的estimate函数即可哈。
我把这个代码放在了码云:https://gitee.com/jmzeng/codes/qbi6023cg9ko5wtsu7hyf52 (大家可以关注一下,如果代码有更新的话,我只会在码云上面更新哈!)而且码云还支持代码搜索功能哦,前往 https://search.gitee.com/ 体验。
当然了,如果你没有安装estimate包是无法使用前面我包装好的函数的。
library(utils)
rforge <- "http://r-forge.r-project.org"
# 对网速有要求哦
install.packages("estimate", repos=rforge, dependencies=TRUE)
library(estimate)
help(package="estimate")
ESTIMATE算法作用于RNA-seq的counts矩阵的合理性
RNA-seq我们在生信技能树应该是至少推出了400篇教程,而且是我们全国巡讲的标准品知识点,其中还有一个阅读量过两万的综述翻译及其细节知识点的补充:
相信大家听完了我B站的RNA-seq分析流程后,对这个数据的应用方向都不陌生。
ESTIMATE就是基于ssGSEA算法,对 stromal and immune 两个基因集在肿瘤表达矩阵的各个样本进行打分。前面我们提到的100篇泛癌研究文献解读之ESTIMATE算法计算肿瘤纯度 ,就已经是把ESTIMATE算法作用于TCGA的全部RNA-seq的counts矩阵了,所以这样做是有理可依的。
其次,既然是ssGSEA算法,大家应该了解它主要是根据基因的排序来进行计算操作,基因的表达量本身并不重要,无论是来自于表达芯片还是来自于RNA-seq,2万个多个基因的排序大体上不会出现冲突。
比如测试数据集的示例是:For Affymetrix platform data, tumor purity are derived from ESTIMATE scores by applying non-linear squares methods to TCGA Affymetrix expression data (n=995).
得到的结果如下:
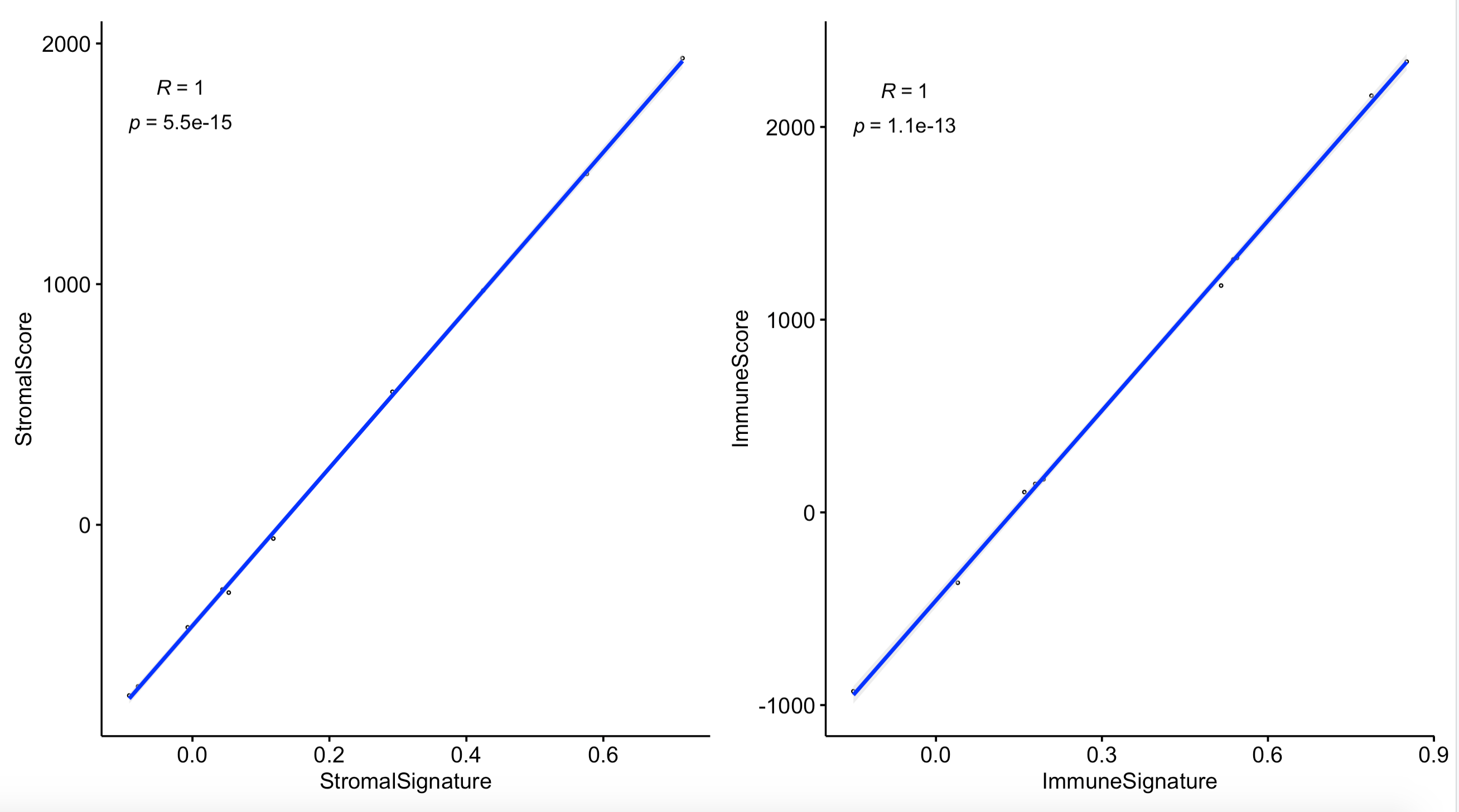
比较ssGSEA算法结果
如果你查看该包的示例数据文件:estimate/extdata/ 就可以看到SI_geneset.gmt基因集文件如下:gse

ssGSEA算法实践我在生信技能树也讲解了不下二十次了,相信粉丝们早就熟练了下面的代码:
OvarianCancerExpr <- system.file("extdata", "sample_input.txt",
package="estimate")
ov=read.table(OvarianCancerExpr)
gmtFile=system.file("extdata", "SI_geneset.gmt",
package="estimate")
library(GSVA)
library(limma)
library(GSEABase)
library(data.table)
geneSet=getGmt(gmtFile,
geneIdType=SymbolIdentifier())
ssgseaScore=gsva(as.matrix(ov), geneSet, method='ssgsea', kcdf='Gaussian', abs.ranking=TRUE)
可以看到这两个基因集走ssGSEA算法后得到基因集打分,跟走estimate得到的打分是高度一致的。
理解了原理,看如何文章都不会盲从!关于gsea,大家可以看我以前在生信技能树的合辑
- GSEA的统计学原理试讲
- 用GSEA来做基因集富集分析
- 批量运行GSEA,命令行版本
- 费九牛二虎之力也无法重现的GSEA图
- GSEA分析合理性讨论
- 做GSEA分析你的基因到底该如何排序
- 重复不出来我费九牛二虎之力重复不出来的GSEA文章?
- 数据挖掘任务-根据前面教程复现ssGSEA热图
文末友情宣传
强烈建议你推荐我们生信技能树给身边的博士后以及年轻生物学PI,帮助他们多一点数据认知,让科研更上一个台阶:
- 生信爆款入门-全球听(买一得五)(第4期),你的生物信息学入门课
- 数据挖掘第2期(两天变三周,实力加量),医学生/临床医师首选技能提高课
- 生信技能树的2019年终总结 ,你的生物信息学成长宝藏
- 2020学习主旋律,B站74小时免费教学视频为你领路,还等什么,看啊!!!
付费内容分割线
为有效杜绝黑粉跟我扯皮,设置一个付费分割线,这样它们就没办法复制粘贴我的代码,也不可能给我留言骂街了!(付费6元,拿到测试数据和代码)
世界顿时清净很多!
综合脚本
链接:https://share.weiyun.com/5qFiXUU 密码:
这个微云链接需要密码,付费6元可以得到,请移步到微信公众号:https://mp.weixin.qq.com/s/UehaaJZgARryH7P25v9wNQ 付费获取哈!