前些天在生信技能树的教程:这样拿差异基因集做肿瘤诊断模型不是明知故问吗,我抛出来了一个开放性问题,也是抨击了一下那个简陋的诊断模型,有粉丝“挑衅”我说如果那个诊断模型不好,有本事我自己发一个好的啊!我就呵呵了,我是否具备抨击某事物的资格并不需要我有能自己创造那个事物的为前提,建议他回去好好学习逻辑学。
但是说到好的诊断模型就不得不提中山大学徐瑞华教授课题组近4年来的系列工作了:
- 2017-05-PNAS:DNA methylation markers for diagnosis and prognosis of common cancers
- 2017-10-nature medicine:Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma
- 2020-01- SCIENCE TRANSLATIONAL MEDICINE : Circulating tumor DNA methylation profiles enable early diagnosis, prognosis prediction, and screening for colorectal cancer
因为他们是中山大学肿瘤防治中心,所以病人队列不用愁,实验设计也合理,集中于挖掘用于各种癌症的诊断和预后模型,都是DNA甲基化位点。我们以最新的结直肠癌为例,看看他们是如何发现可用于液体活检的特定ctDNA甲基化位点,而且是诊断和预后模型一起搞!第一阶段:数据挖掘
首先需要下载TCGA的结直肠癌甲基化位点信号矩阵文件:
- Tissue DNA methylation data were obtained from the TCGA (TCGA, TCGA-COAD, and TCGA-READ).
还需要正常人的血液的甲基化信号值作为对照: - Whole-blood DNA methylation profiles from healthy donors were generated in an aging study (GSE40279)
上面的两个队列是为了确定直肠癌特异性甲基化位点,做的是差异分析,确定了 top 1000 methylation markers,如果你感兴趣具体差异分析的统计学算法,以及差异分析的热图可视化结果建议看原文:Fig. S8. Unsupervised hierarchical clustering of the top 1000 methylation markers differentially methylated between CRC tumor DNA and normal blood.
如果你感兴趣他们挑选的位点,可以去看Fig. S1. List of methylation correlated blocks used for cd-score generation.
然后在自己医院收集的液体活检的ctDNA甲基化队列就无需使用450K或者850K芯片啦,毕竟一个450K或者850K芯片起码3000块钱,如果是自己搞2000个样本,6百万就砸进去了。使用已经确定好的直肠癌特异性甲基化位点,成本锐减到之前的十分之一!

用来构建诊断和预后模型的液体活检的ctDNA甲基化队列(主要的数据分析基于此) - The cfDNA cohort consisted of 801 patients with CRC and 1021 healthy controls.
- 这些样本无需450K或者850K芯片,使用的是 capture-sequencing cfDNA。
- NA methylation rate at each MCB was determined using deep sequencing of bis-DNA captured with molecular inversion probes.
因为这个文章是诊断和预后模型一起做,所以数据分析环节工作量比较大,大体上的流程如下所示:
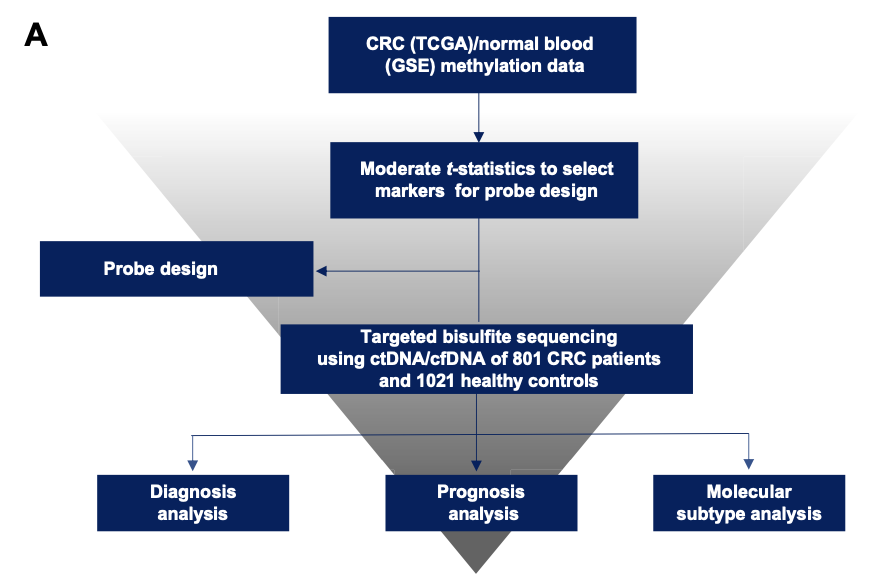
为了方便讲解,我们仅仅是拿疾病诊断模型来介绍,毕竟我们的重心是教程:这样拿差异基因集做肿瘤诊断模型不是明知故问吗。疾病诊断模型构建流程
需要提前理解 combined prognosis score (cp-score) 概念,以及UniCox and LASSO-Cox,还有随机森林的算法实现。
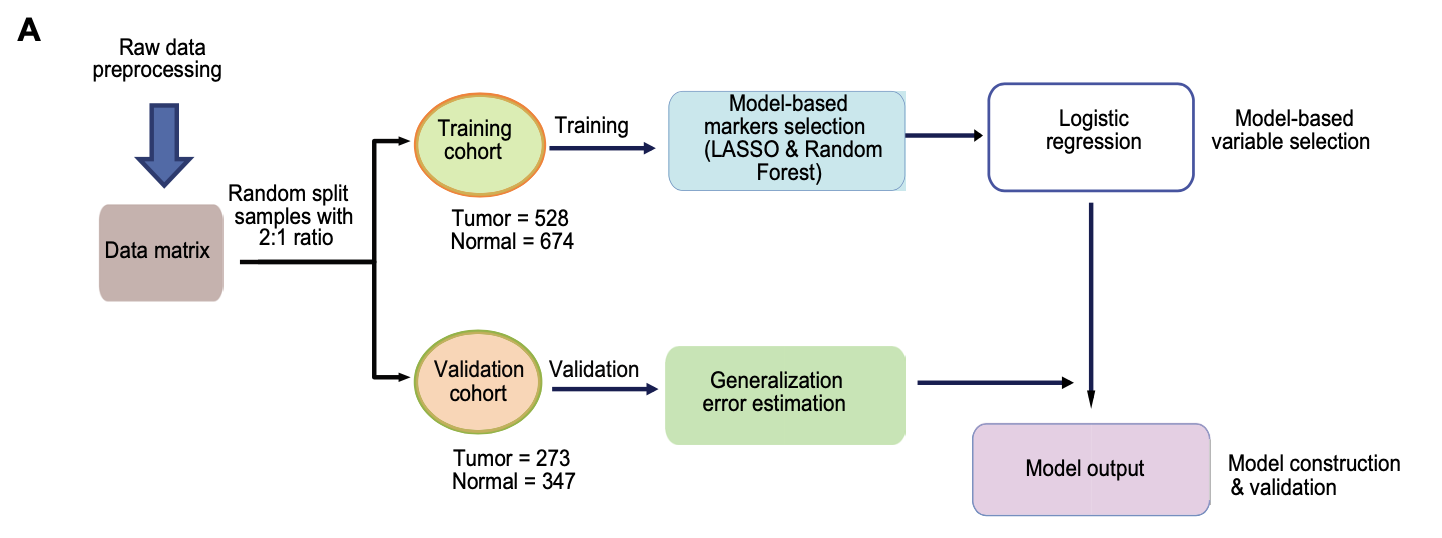
一定要注意拆分数据集; - The cfDNA methylation data from this cohort were randomly divided into training and validation datasets with a 2:1 ratio to build both diagnostic and prognostic models.
不过,值得注意的是这个时候数据建模其实就很有意思了,因为文章附带的原始数据集并不是公开的,就是近1000个甲基化位点在近2000样本的信号值矩阵文件拿不到,所以我们没办法重复出来这个分析过程的。训练数据集
前面的公共数据库挖掘,重要就是差异分析确定了直肠癌特异性甲基化位点,从几十万个位点减少到了一千个这样的数量级。(这个步骤都是公共数据可以下载,然后复现它)
然后病人的ctDNA甲基化信号值矩阵再次使用lasso和随机森林,进行减少到只剩下9个甲基化位点。(这个时候,缺乏近1000个甲基化位点在近2000样本的信号值矩阵文件,所以不能复现)
如下所示的热图很清晰展现了这9个甲基化位点在cancer和normal组别的信号值差异情况,当然了,如果能横向z-score一下也许会更明显,不过z-score的也有弊端,就是失去了甲基化探针互相之间的信号值高低信息。
可以看到,这9个甲基化位点组成的疾病诊断模型的AUC值也是超级高,但是和前面我们介绍的:这样拿差异基因集做肿瘤诊断模型不是明知故问吗,不是一回事,右边的混淆矩阵也很清晰的看出来了模型的效果。
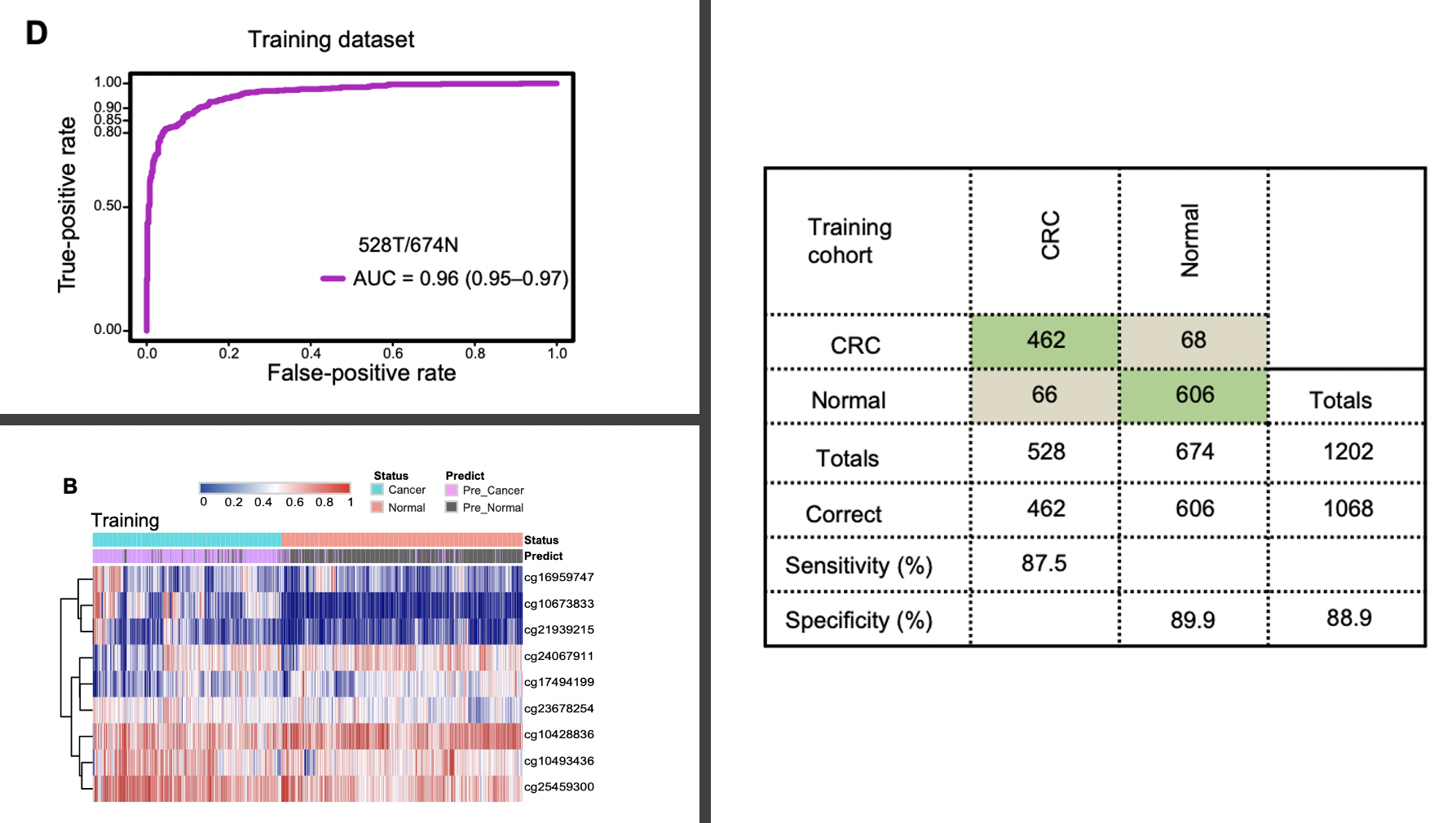
比较重要的一点就是样本队列的大小,另外一个优点就是拆分了测试集和验证集。验证数据集
因为缺乏近1000个甲基化位点在近2000样本的信号值矩阵文件,所以我们只能是继续看图,前面的测试数据集找到的9个甲基化位点建立好了模型,在这个验证数据集里面效果如下:
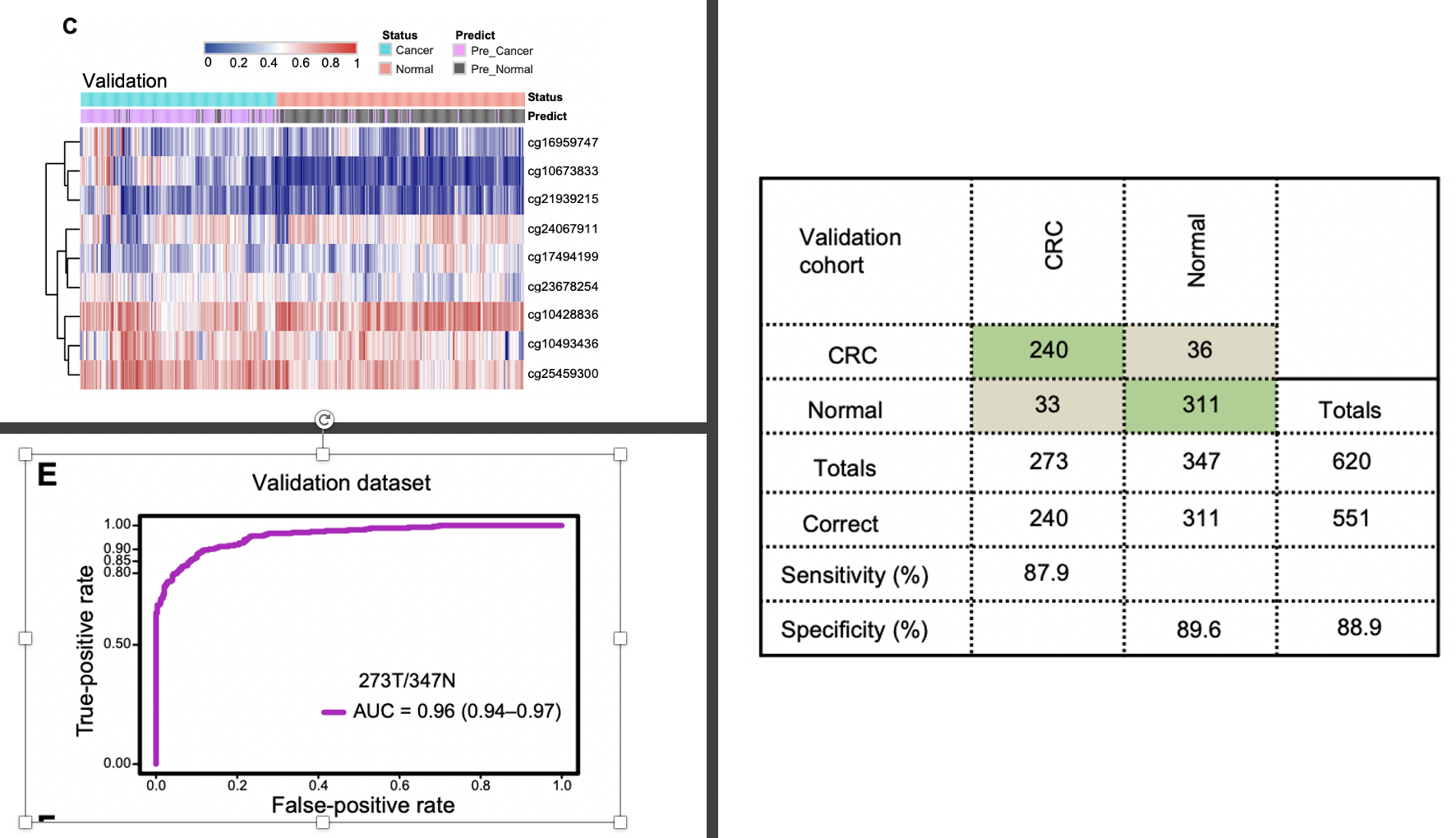
如果仅仅是把病人队列区分成为测试数据集和验证数据集,然后进行内部多次建模挑选最好的发表,其实就成为了数据游戏了。但是中山大学肿瘤防治中心,所以病人队列不用愁,继续扩大样本量做人群队列研究。
值得注意的是,做大规模人群筛选,需要越觉得越好,所以研究者从前面的9个甲基化位点构建的诊断模型里面又定位到了一个表现最好的,就是 cg10673833第二阶段:人群队列验证
背靠医院资源,大规模人群队列是可以实现的:
- The prospective CRC screening cohort was composed of a total of 16,890 subjects, aged between 45 and 75 years and without CRC- related symptoms, who participated in this study between January 2015 and December 2017.
可以看到,不到3年时间就收集了近2万人,这个执行力,佩服!而且 Clinical characteristics of all participants from this cohort are listed in table S2,大家感兴趣可以去看看医院是如何记录病人临床信息的。而且招募的受试者需要符合以下条件 - (i) men and women,
- (ii) age 45 to 75 years,
- (iii) never diagnosed with cancer,
- (iv) consent to receive and complete investigation questionnaire,
- (v) able and willing to undergo a screening colonoscopy within 90 days of enrollment,
- (vi) able and willing to provide plasma samples.
流程图如下:
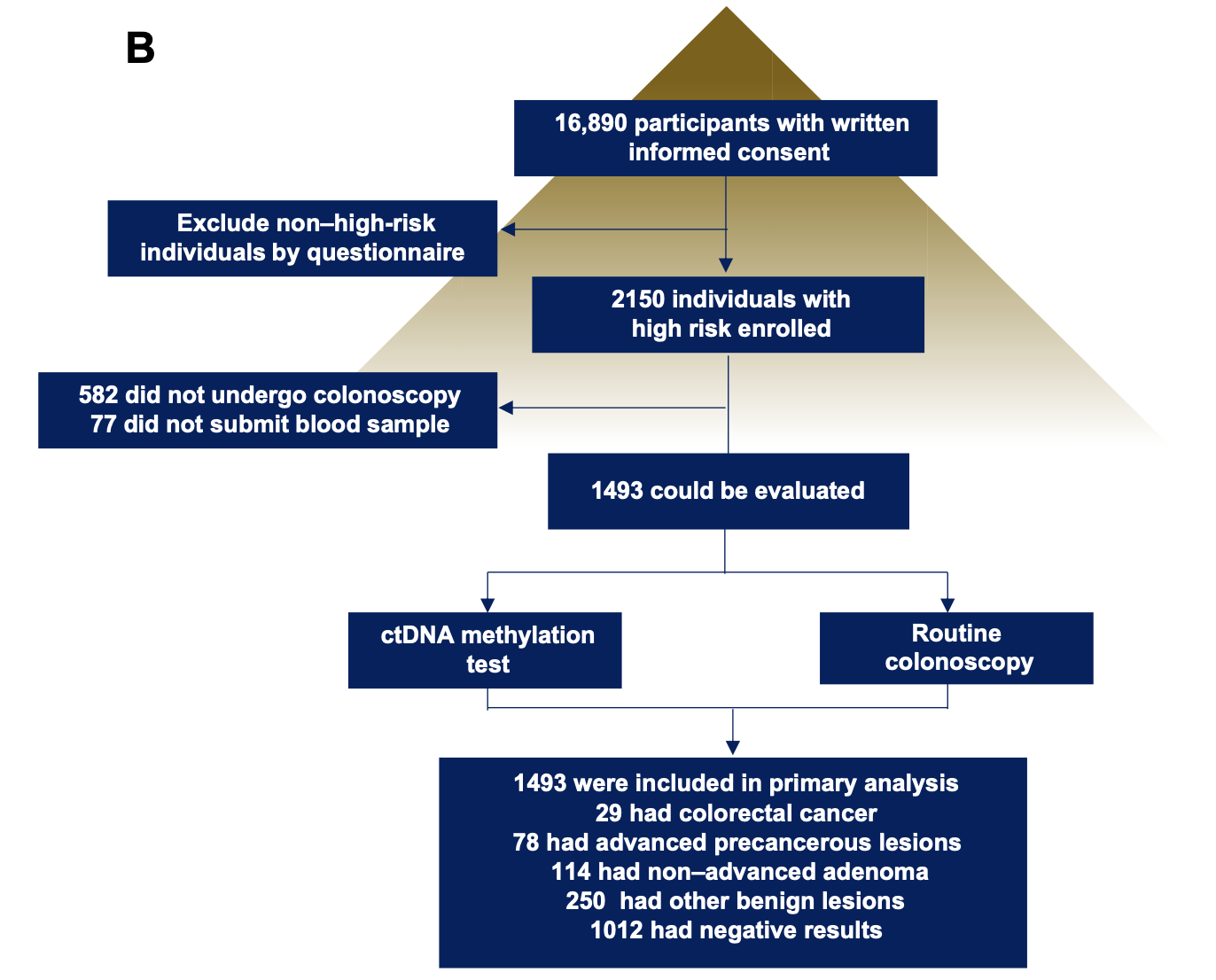
可以看到, 根据研究者建立的模型,可以比较好的在人群队列发挥针对和预后判断的效果。
总之,研究者提出依据一个新发现的甲基化标志性位点cg10673833来简化癌症筛查的可能性,敏感性为89.7%,特异性为86.8%,说明能够通过血浆中cg10673833甲基化标志物准确预测癌前病变。
值得注意是cg10673833这个甲基化位点虽然identified 26 of 29 participants with CRC,但是For advanced precancerous lesions, the sensitivity was 33.3%。The cg10673833 marker performed less well for precancerous lesions, as would be expected; however, in this circumstance, it might be beneficial to use more than just one marker. Our results indicated that cg10673833 was superior to other currently reported cfDNA methylation markers for CRC screening.
另外的几个研究类似,大家可以自行搜索关注中山大学徐瑞华教授课题组近4年来的系列工作了: - 2017-05-PNAS:DNA methylation markers for diagnosis and prognosis of common cancers
- 2017-10-nature medicine:Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma
- 2020-01- SCIENCE TRANSLATIONAL MEDICINE : Circulating tumor DNA methylation profiles enable early diagnosis, prognosis prediction, and screening for colorectal cancer
也可以考虑直接去他们课题组或者中山大学肿瘤防治中心参与这样的研究!
如果你是生信工程师,那么我不得不强推一下中山大学肿瘤防治中心生物信息平台给你!关于中山大学肿瘤防治中心生物信息平台
中山大学肿瘤防治中心生物信息平台成立于2016年,地址位于中心2号楼20楼。作为精准医疗平台的重要组成部分,平台致力于为中心的基础和临床研究提供肿瘤学相关的生物信息支撑,并独立开展肿瘤相关课题。平台可提供高通量测序、基因芯片、质谱等大数据的处理分析以及临床医学相关统计建模等技术咨询和课题合作。硬件设施方面,平台依托“天河二号”超级计算机搭建了基于超算的生物信息分析平台;另一方面购置并部署了存储超过2PB、总CPU超过200个的集群、20+工作站等计算设备。生物信息平台将为中心的肿瘤精准医疗研究和应用提供高效、快速的生物信息资源和技术支撑。
文末友情推荐
要想真正入门生物信息学建议务必购买全套书籍,一点一滴攻克计算机基础知识,书单在:什么,生信入门全套书籍仅需160 。
如果大家没有时间自行慢慢摸索着学习,可以考虑我们生信技能树官方举办的学习班: - 数据挖掘学习班第4期(线上直播3周,马拉松式陪伴,带你入门)
- 生信爆款入门-第6期(线上直播4周,马拉松式陪伴,带你入门)
如果你没有服务器的话,做NGS数据分析实战可能会有点勉强,建议考虑:每天不足一块钱,定制生信云送给你