一般来说,数据分析的任何一个点都会有成百上千个实验室在为之努力,比如比对,定量,可变剪切,融合基因这些步骤,那么同时也会有很多工具测评的文章,比如针对融合基因的随便搜一下,就可以发现下面两个:
- 2016的文章:Comparative assessment of methods for the fusion transcripts detection from RNA-Seq data 测评了12个工具。
- 2019的文章:Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods 测评了23个工具算法。
大家完全可以直接快速浏览这样的文章就能迅速了解这个领域了!
融合基因算法的两个流派
转录组拼接来探索新的融合基因情况
2017年BMC文章:De novo assembly and characterization of breast cancer transcriptomes identifies large numbers of novel fusion-gene transcripts of potential functional significance, 通过对乳腺癌的转录组数据进行拼接来探索新的融合基因情况。
作者开发了流程来做the de novo assembly and characterization of chimeric transcripts in 55 primary breast cancer and normal tissue samples. 这里,作者从TCGA数据库里面选取45个乳腺原位癌以及10个正常样品测序的原始数据,走下面的步骤:
- 先用Trim Galore做质控。
- 然后用ABySS来进行转录本组装(多个kmer值同时组装),组装好的contig先用RepeatMasker把ployA尾巴屏蔽掉。
- 然后用blat跟参考基因组比对,BLAT产生的pslx文件可以直接作为R-SAP流程的输入文件,来寻找 chimeric transcript ,对找到的chimeric transcript结果进行一系列的严格过滤。
- 再用bowtie把原始的转录组测序数据比对到找到的chimeric transcript序列,最后得到了1959个chimeric transcript序列。
仅关注 split reads and discordant read pairs
比如工具FusionScan的文章:FusionScan: accurate prediction of fusion genes from RNA-Seq data 里面的流程图:

可以看到全程依赖于比对环节,提取 split reads and discordant read pairs。
融合基因的数据挖掘思路
融合基因本身是肿瘤治疗的靶点,所以可以看有无某融合事件进行分组后看生存差异,以及一系列分组后的标准分析。
大量的NGS队列研究都仅仅是分析到了表达量层面,而且提供测序原始数据的,所以可以下载原始数据去看融合基因突变全景图,以及对应的生物学意义!比如文章:Transcriptome analysis offers a comprehensive illustration of the genetic background of pediatric acute myeloid leukemia 就是专注解释了一个AML队列的融合基因情况:
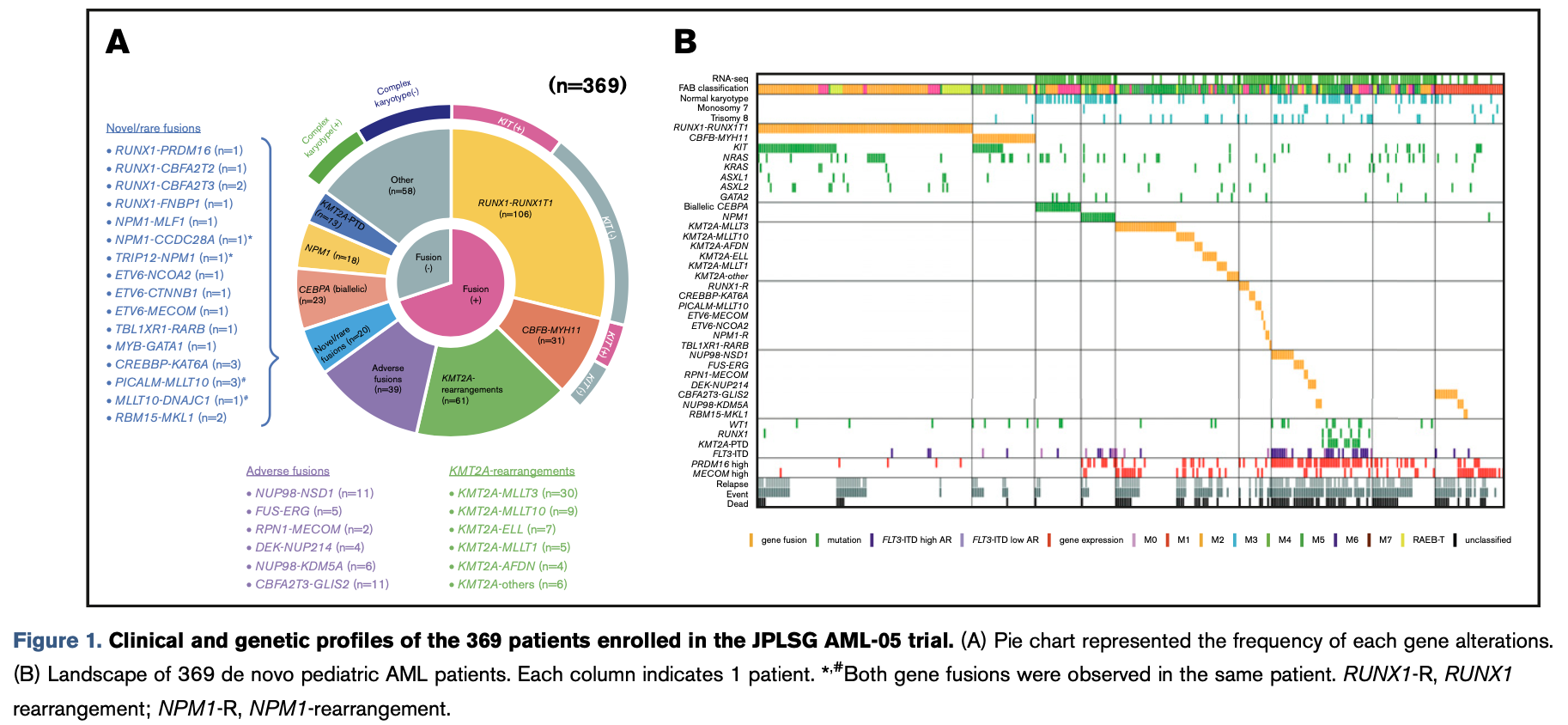
TCGA的融合基因
在数据库网页工具:https://tumorfusions.org/ 可以下载和查询针对TCGA的RNA-seq数据的全部基因融合事件,全称是:TUMOR FUSION GENE DATA PORTAL
同时还有一个:ChimerDB 4.0: an updated and expanded database of fusion genes 也提供查询TCGA的RNA-seq数据的基因融合事件
以及 https://ccsm.uth.edu/FusionGDB/index.html
融合基因的临床应用前景
这个很有意思,之前听瑞金医院的朋友提到过,准备把RNA-seq的临检常规化,我特意搜索了一下,发现已经有一些RNA-seq的融合基因panel。具体推荐大家看2个文章:
- Diagnosis of fusion genes using targeted RNA sequencing
- Development and Verification of an RNA Sequencing (RNA-Seq) Assay for the Detection of Gene Fusions in Tumors
三代测序会变革融合基因领域
在三代测序的超长reads的优点面前,以前那些在ngs的短reads的算法难题都不再是问题,所以我还是蛮相信三代测序会变革融合基因领域,大家可以去搜索一些文献,比如:A Nanopore Sequencing–Based Assay for Rapid Detection of Gene Fusions
因为三代测序不是我的领域,我就先不总结了,也许三五年后我会回过头来继续这方面知识整理吧,那个时候我在生物信息学领域就达到了10年分享的小目标!
不过恰好有一个三代测序的研究实验室有博士后招聘,博士后训练机会,俄亥俄州立大学,生物医学信息系:
统计生物信息学实验室(http://augroup.org/)主要关注开发和应用生物信息学方法在高通量的测序数据 分析上,特别是三代测序的数据分析。


癌症研究领域里面三代测序肯定会发光发热,而且走融合基因这一个小领域也不错,感兴趣的博士们赶快联系吧!
融合基因算法工具列表
其实说了那么多,大家想要的就是一个好用的工具,去把RNA-seq数据鉴定一些融合基因事件。但是工具有近百个之多!
我在 https://www.biostars.org/p/45986/ 看到一个精彩的回答里面,他们总结了从RNA-seq数据里面鉴定融合基因的工具列表:
- 1. Barnacle
- 2. Bellerophontes
- 3. BreakDancer
- 4. BreakFusion
- 5. BreakPointer
- 6. ChimeraScan
- 7. Comrad
- 8. CRAC
- 9. deFuse
- 10. Dissect
- 11. EBARDenovo
- 12. EricScript
- 13. FusionAnalyser
- 14. FusionCatcher
- 15. FusionFinder
- 16. FusionHunter
- 17. FusionMap
- 18. FusionQ
- 19. FusionSeq
- 20. IDP-fusion
- 21. iFUSE
- 22. InFusion
- 23. INTEGRATE
- 24. JAFFA
- 25. LifeScope
- 26. MapSplice
- 27. MOJO
- 28. nFuse
- 29. Pegasus
- 30. PRADA
- 31. ShortFuse
- 32. SnowShoes-FTD
- 33. SOAPFuse
- 34. SOAPFusion
- 35. STAR
- 36. STAR-Fusion
- 37. TopHat-Fusion
- 38. TRUP
- 39. ViralFusionSeq
文末友情推荐
要想真正入门生物信息学建议务必购买全套书籍,一点一滴攻克计算机基础知识,书单在:什么,生信入门全套书籍仅需160 。
如果大家没有时间自行慢慢摸索着学习,可以考虑我们生信技能树官方举办的学习班:
如果你没有服务器的话,做NGS数据分析实战可能会有点勉强,建议考虑:每天不足一块钱,定制生信云送给你